如何处理矫正GEO数据批次效应(batch effect)
在GEO数据挖掘分析中经常会遇到会遇到样品数据不足,需要联合分析多个芯片数据进行分析,那么能将这些数据直接混合分析吗?如果贸然混合,会有什么问题?这个问题就是batch effect。
什么是batch effect?
不同平台的数据,同一平台的不同时期的数据,同一个样品不同试剂的数据,以及同一个样品不同时间的数据等等都会产生一种batch effect 。这种影响如果广泛存在应该被足够重视,否则会导致整个实验和最终的结论失败。
我简单说下什么叫做batch effect。比对实验组和对照组,不同的处理是患病和不患病(测序时,先测得疾病,然后测得正常),然后你通过分析,得到很多差异表达的基因。现在问题来了,这个差异表达的结果是和你要研究的因素有关,还是时间有关,这个问题里时间就会成为干扰实验结果的因素,这个效应就是batch effect。
如何检测是否存在这种效应呢
最简单的就是记录实验中时间这个变量,然后对差异表达的基因进行聚类,看是否都和时间相关,如果相关就证明存在batch effect。
同样,如果不同平台的数据之间存在batch effect ,就不能简单的整合。
大家可能都会问标准化,会不会处理掉batch effect ?
答案是能够减弱,不能从根本上消除。如下图,b是a进行过标准化的结果,从样本上看都一直,没有什么问题,但是落实到基因层面,c图中还是有明显的batch effect,d图中通过时间进行聚类,很明显可以看出差异表达主要是由于时间引起的。
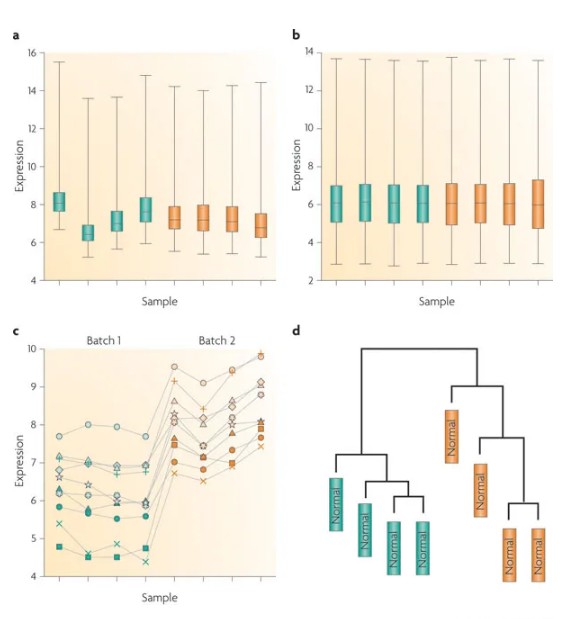
矫正批次效应
假如解决了这个批次问题,不仅可以让实验更可靠,更厉害的是,我们可以做多个芯片的联合分析了。矫正批次效应有两种方法:
1.使用sva的中combat包来校正批次效应
下面是举例子: 安装必要的R包并加载,comat就在sva包中。
# 安装包,提前添加镜像,加快安装速度
if (!requireNamespace("BiocManager", quietly=TRUE)){
install.packages("BiocManager")
}
options(BioC_mirror="https://mirrors.tuna.tsinghua.edu.cn/bioconductor")
local({r <- getOption("repos")
r["CRAN"] <- "http://mirrors.tuna.tsinghua.edu.cn/CRAN/"
options(repos=r)})
BiocManager::install("sva")
BiocManager::install("bladderbatch")
library(sva)
library(bladderbatch)
library('Biobase')
library('GEOquery')
data(bladderdata)
#bladder 的属性是EsetExpressionSet,所以可以用pData和exprs方法
pheno <- pData(bladderEset) # 注释信息
edata <- exprs(bladderEset) # 表达矩阵
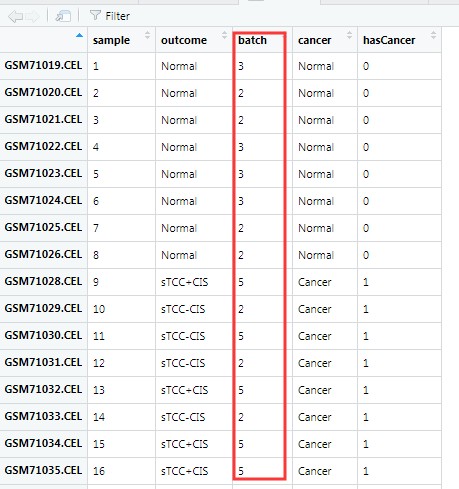
看一下pheno里面有54行,4列构成,里面记录了批次信息
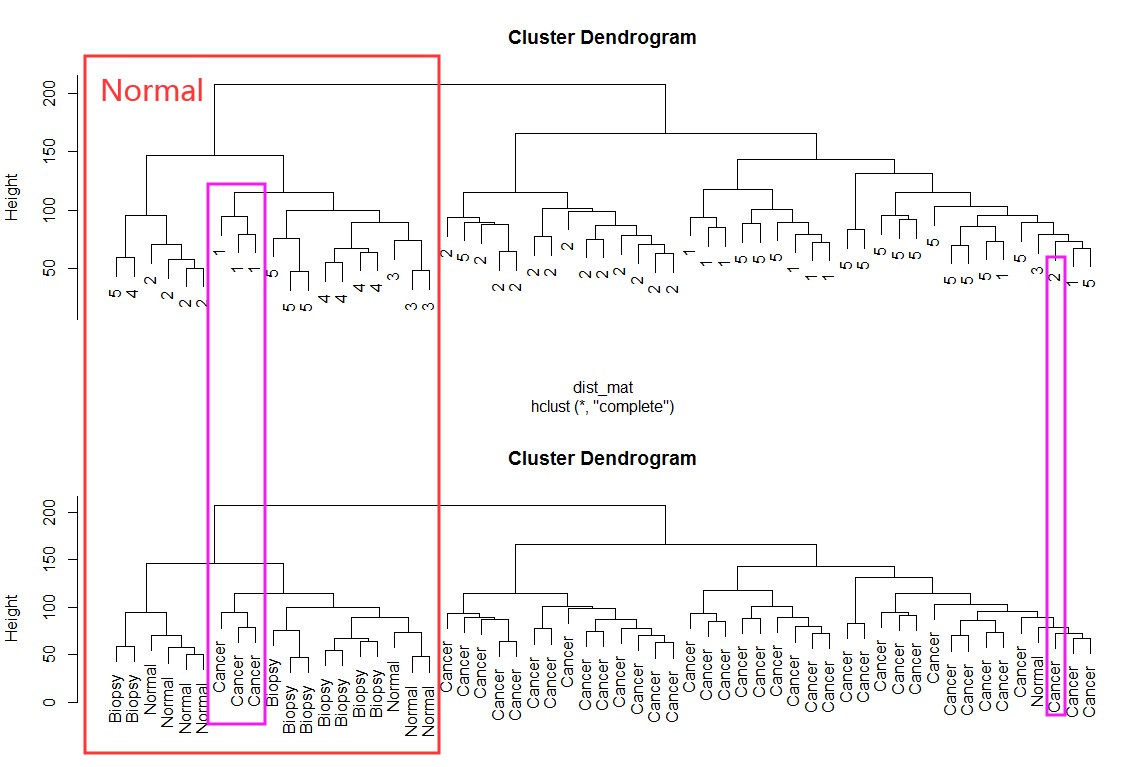
有没有批次效应可以通过使用Hierarchical clustering的聚类方法去看一下聚类的情况:例如下面数据中,批次1中cancer样品与normal有混合,需要矫正一下:
dist_mat <- dist(t(edata))
clustering <- hclust(dist_mat, method = "complete")
par(mfrow=c(2,1))
plot(clustering, labels = pheno$batch)
plot(clustering, labels = pheno$cancer)
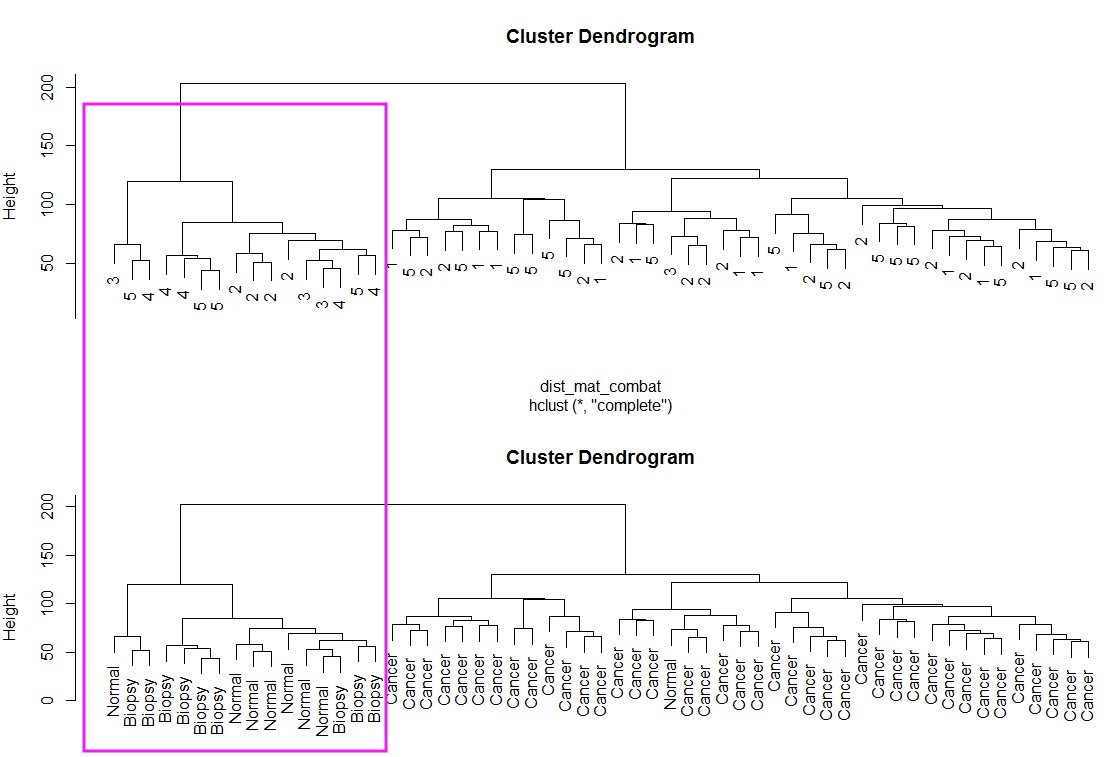
 校正批次效应:model可以有也可以没有,如果有,也就是告诉combat,有些分组本来就有差别,不要给我矫枉过正!
校正批次效应:model可以有也可以没有,如果有,也就是告诉combat,有些分组本来就有差别,不要给我矫枉过正!
#再做一个分组列,用于批次效应中排除项。
pheno$hasCancer <- as.numeric(pheno$cancer == "Cancer")
#分组模型
model <- model.matrix(~hasCancer, data = pheno)
combat_edata <- ComBat(dat = edata, batch = pheno$batch, mod = model)
dist_mat_combat <- dist(t(combat_edata))
clustering_combat <- hclust(dist_mat_combat, method = "complete")
par(mfrow=c(2,1))
plot(clustering_combat, labels = pheno$batch)
plot(clustering_combat, labels = pheno$cancer)
2.使用 limma 的 removeBatchEffect 函数
还是利用上面的数据利用李
dat = edata, batch = pheno$batch
library("limma")
design <- model.matrix(~pheno$batch+pheno$hasCancer)
limma.edata <- removeBatchEffect(edata,
batch = pheno$batch,
design = design)
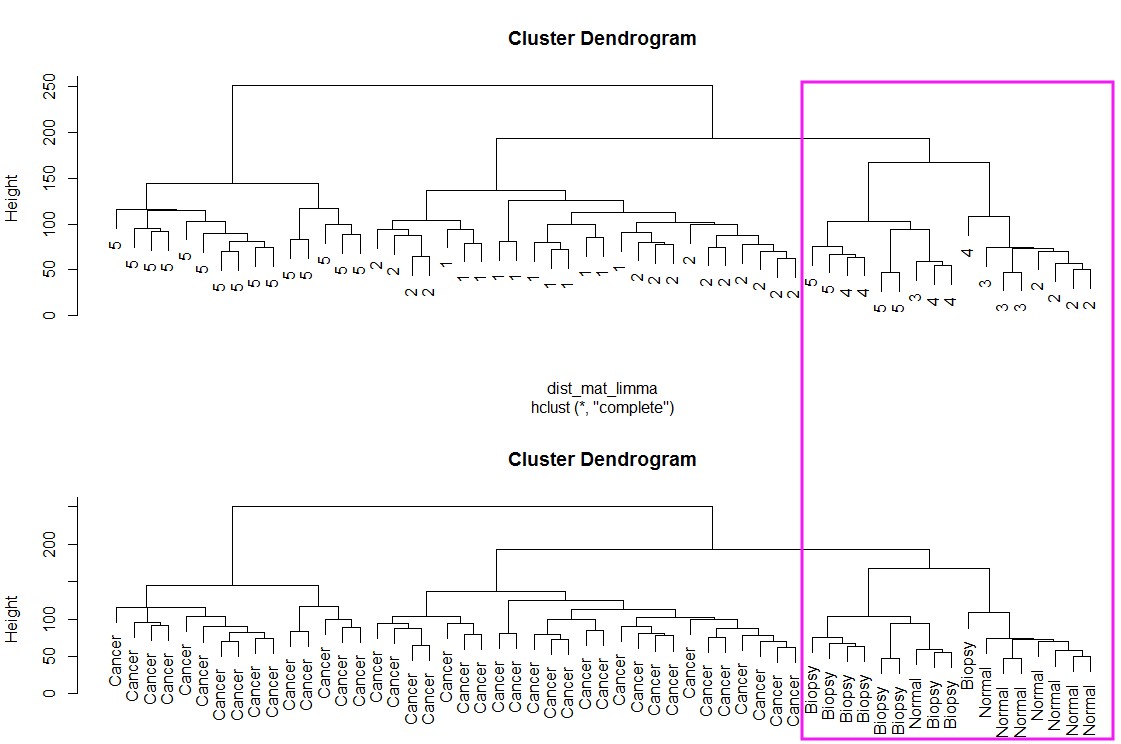
dist_mat_limma <- dist(t(limma.edata))
clustering_limma <- hclust(dist_mat_limma, method = "complete")
par(mfrow=c(2,1))
plot(clustering_limma, labels = pheno$batch)
plot(clustering_limma, labels = pheno$cancer)
参考文献:https://www.nature.com/articles/nrg2825?report=reader
更多生物信息课程:
1. 文章越来越难发?是你没发现新思路,基因家族分析发2-4分文章简单快速,学习链接:基因家族分析实操课程、基因家族文献思路解读
2. 转录组数据理解不深入?图表看不懂?点击链接学习深入解读数据结果文件,学习链接:转录组(有参)结果解读;转录组(无参)结果解读
3. 转录组数据深入挖掘技能-WGCNA,提升你的文章档次,学习链接:WGCNA-加权基因共表达网络分析
4. 转录组数据怎么挖掘?学习链接:转录组标准分析后的数据挖掘、转录组文献解读
5. 微生物16S/ITS/18S分析原理及结果解读、OTU网络图绘制、cytoscape与网络图绘制课程
6. 生物信息入门到精通必修基础课:linux系统使用、biolinux搭建生物信息分析环境、linux命令处理生物大数据、perl入门到精通、perl语言高级、R语言画图、R语言快速入门与提高、python语言入门到精通
7. 医学相关数据挖掘课程,不用做实验也能发文章:TCGA-差异基因分析、GEO芯片数据挖掘、 GEO芯片数据不同平台标准化 、GSEA富集分析课程、TCGA临床数据生存分析、TCGA-转录因子分析、TCGA-ceRNA调控网络分析
8.其他,二代测序转录组数据自主分析、NCBI数据上传、二代fastq测序数据解读、
9.全部课程可点击:组学大讲堂视频课程
- 发表于 2019-11-19 15:45
- 阅读 ( 27421 )
- 分类:GEO
