BIOM 微生物数据格式及文件转换
BIOM 微生物数据格式
扩增子分析视频课程推荐:https://study.omicsclass.com/index
BIOM格式是微生物组领域最常用的结果保存格式,优点是可将OTU或Feature表、样本属性、物种信息等多个表保存于同一个文件中,且格式统一,体积更小巧,目前被微生物组领域几乎所有主流软件所支持

BIOM目前分为1.0 JSON和2.0 HDF5两个版本
1.0 JSON是编程语言广泛支持的格式,类似于散列的键值对结果。会根据数据松散程度,选择不同的存储结构来节省空间。
2.0 HDF5是二进制格式,被许多程序语言支持,读取更高效和节约空间。
如何节约存储:
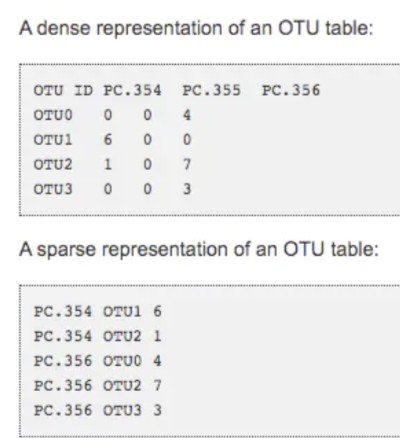
- biom格式转换常用命令:
- 转换经典表格为HDF5或JSON格式
biom convert -i table.txt -o table.from_txt_json.biom --table-type="OTU table" --to-json biom convert -i table.txt -o table.from_txt_hdf5.biom --table-type="OTU table" --to-hdf5
- 转换biom为经典格式
biom convert -i table.biom -o table.from_biom.txt --to-tsv
- 转换biom为经典格式,并在最后列包括物种注释信息
biom convert -i table.biom -o table.from_biom_w_taxonomy.txt --to-tsv --header-key taxonomy
- 转换biom为经典格式,并在最后列包括物种注释信息,并改名为ConsensusLineage
- 此功能对于一些软件要求指定的列名有很有用。
- biom convert -i table.biom -o table.from_biom_w_consensuslineage.txt --to-tsv --header-key taxonomy --output-metadata-id "ConsensusLineage"
- 带物种注释表格互转
biom convert -i table.biom -o table_tax.txt --to-tsv --header-key taxonomy biom convert -i table_tax.txt -o new_table.biom --to-hdf5 --table-type="OTU table" --process-obs-metadata taxonomy biom convert -i table_tax.txt -o new_table.biom --to-json --table-type="OTU table" --process-obs-metadata taxonomy
取子集亚组进行分析;
biom subset-table -i otu_table.biom -a sample -s samples_list.txt -o otu_table_subset.biom
biom文件中过滤方法:
如果系统中安装了qiime1 ,会有关于biom文件得过滤得脚本,方便我们筛选自己得数用于后续分析:
# 按样品数据测序量过滤:选择counts>30000的样品
filter_samples_from_otu_table.py -i otu_table.biom -o otu_table1.biom -n 30000
# 查看过滤后结果:
biom summarize-table -i otu_table1.biom
# 按样品数据测序量过滤:选择counts<10000的样品
filter_samples_from_otu_table.py -i otu_table.biom -o otu_table_no_high_coverage_samples.biom -x 10000
# 按OTU丰度过滤:选择相对丰度均值大于十万分之一的OTU
filter_otus_from_otu_table.py --min_count_fraction 0.00001 -i otu_table.biom -o otu_table1.biom
# 按物种过滤OTU表:去除p__Chloroflexi菌门等
filter_taxa_from_otu_table.py -i otu_table.biom -o otu_table1.biom
#Split otu_table.biom into per-study OTU tables, and store the results in ./per_study_otu_tables/
split_otu_table.py -i otu_table.biom -m Fasting_Map.txt -f Treatment -o per_study_otu_tables
#Split otu_table.biom into multiple biom tables based on the Treatment and Color of the samples
split_otu_table.py -i otu_table.biom -m Fasting_Map.txt -f Treatment,Color -o ./per_study_otu_tables/
- 发表于 2020-08-17 11:20
- 阅读 ( 11129 )
- 分类:宏基因组
