重测序群体遗传进化分析之—进化树构建
重测序大家都不陌生,它是检测样本基因组变异(SNP,indel,SV,CNV)的主要手段之一,有了这些变异信息,后续可以做很多分析工作,例如: 遗传群体可以进行遗传图谱构建、BSA分析等;大样本量自然群体,可以进行群体遗传进化分析等。今天,小编先给大家群体遗传进化分析内容中的进化树构建;教您如何利用重测序获得的SNP数据构建系统进化树,关于进化树的构建原理见:进化树构建原理。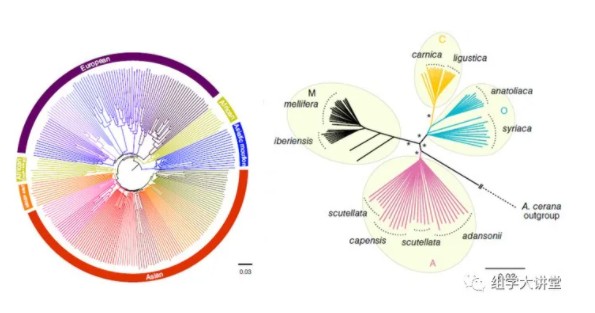
构建进化树的软件
构建进化树的方法和软件很多,前面我们讲解构建进化树的原理时提过,最准确的方法为贝叶斯方法,但是贝叶斯的方法计算量太大太耗时,对于大量的数据不适用,其次就是最大似然法,这里我们解释三种利用最大似然法构建进化树的软件:fasttree,iqtree2,RAxML-ng.输入的数据为做群体遗传进化得到的SNP数据。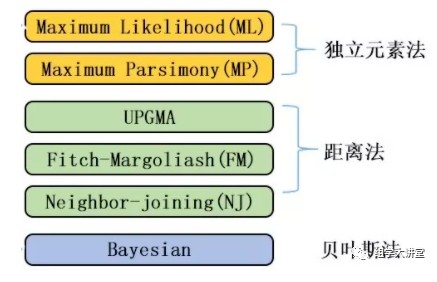
数据准备做完重测序分析后,我们可以得到包含 SNP的变异结果vcf文件作为输入文件。详见课程:重测序数据分析课程,然后利用omicsclass/pop-evol-gwas:v1.3 镜像进行前期数据准备与后续进化树构建分析:
#运行环境准备:docker镜像启动
#镜像下载:
docker pull omicsclass/pop-evol-gwas:v1.3
#启动遗传进化镜像
docker run --rm -it -m 4G --cpus 1 -v D:\pop:/work omicsclass/pop-evol-gwas:v1.3
#对vcf文件进行数据过滤
vcftools --gzvcf all.varFilter.vcf.gz --recode --recode-INFO-all --stdout --maf 0.05 --max-missing 0.7 --minDP 4 --maxDP 1000 \
--minQ 30 --minGQ 0 --min-alleles 2 --max-alleles 2 --remove-indels |gzip - > clean.vcf.gz
#利用tassel软件对文件进行排序
run_pipeline.pl -Xmx30G -SortGenotypeFilePlugin -inputFile clean.vcf.gz \
-outputFile clean.sorted.vcf.gz -fileType VCF
#vcf文件格式转换成Phylip格式,用于后续构建进化树
run_pipeline.pl -Xmx5G -importGuess $workdir/00.filter/clean.sorted.vcf.gz \
-ExportPlugin -saveAs supergene.phy -format Phylip_Inter
1. FastTree 构建进化树
FastTree 是基于最大似然法构建进化树的软件,它最大的特点就是运行速度快,支持几百万条序列的建树任务。但是fasttree不支持bootstrap检验以及支持的替换模型有限。官网如下:http://www.microbesonline.org/fasttree/
替换模型选择
FastTree 支持核酸和蛋白的进化树构建,对于核酸,可选的替换模型包括以下几种:JC(Jukes-Cantor)、GTR(generalized time-reversible),默认的模型为JC。对于蛋白质,可选的替换模型包括以下几种:JTT (Jones-Taylor-Thornton 1992)、LG(Le and Gascuel 2008)、WAG(Whelan & Goldman 2001) 默认的模型为JTT。FastTree要求输入的多序列比对结果为FASTA或者Phylip格式。
构建进化树命令举例:
fasttree -nt -gtr supergene.fa > fasttree.nwk
FastTree 其他命令举例:
#对于蛋白质的进化树构建,基本用法如下
fasttree protein.fasta > tree
#也可以选择LG或者WAG替换模型,用法如下
fasttree -lg protein.fasta > tree
fasttree -wag protein.fasta > tree
#对于核酸序列,基本用法如下
fasttree -nt nucleotide.fasta > tree
#也可以选择GTR替换模型,用法如下
fasttree -nt -gtr nucleotide.fasta > tree
2. IQ-tree 构建进化树
IQ-tree也是一款最大似然法构建进化树的软件,目前IQ-tree已经更新到2.0版本功能和性能也有很大的提升,主要有四大功能,高效建树(efficient tree reconstruction),模型选择(modelfinder: fast and accurate model selection),超快bootstrap(ultrafast bootstrap approximation),大型数据(big data analysis),以上特点特别适用于高通量测序的大量SNP构建进化树。
模型选择
构建进化树有很多模型对于初学者往往不知道那种模型最适合,iqtree提供自动模型选择功能,使用的软件为modelfinder。Modelfinder是一款速度超快的自动最佳模型选择软件。其在保证准确性的情况下,速度上比jmodeltest(针对DNA)和prottest(针对蛋白)要快100倍(ModelFinder is up to 100 times faster than jModelTest/ProtTest.),使用命令举例:
#自动选择最佳模型并构建进化树:-m MFP
iqtree -s supergene.phy -m MFP
#只是想找最佳模型不构建进化树:
iqtree -s example.phy -m MF
#查找模型计算过程:
ModelFinder will test up to 546 protein models (sample size: 36415) ...
No. Model -LnL df AIC AICc BIC
1 LG 10134094.366 349 20268886.731 20268893.505 20271854.186
2 LG+I 10133927.677 350 20268555.354 20268562.167 20271531.312
3 LG+G4 10043239.052 350 20087178.104 20087184.917 20090154.062
4 LG+I+G4 10043175.024 351 20087052.048 20087058.900 20090036.508
5 LG+R2 10063911.721 351 20128525.442 20128532.294 20131509.902
6 LG+R3 10045448.117 353 20091602.235 20091609.165 20094603.701
MFP为ModelFinder Plus的缩写,该参数使程序执行ModelFinder选择最适模型并完成建树分析。ModelFinder为许多不同的模型计算初始简约树的逻辑概率,并产生Akaike information criterion (AIC),* corrected Akaike information criterion* (AICc), and* the Bayesian information criterion* (BIC)三个结果标准值,通常ModelFinder选择BIC分数最低的模型(当然也可以指定AIC和AICc通过指定选项-AIC或者-AICc)。如果你想节约时间,可指定选择的模型及编码参数,例如:从WAG ,LG ,JTT 核苷酸替代模型里选一个: -mset WAG,LG,JTT;在+G和+I,以及+I+G三个里面选择rate :-mrate G,I,I+G;heterogeneity参数:-mfreq FU,F命令行如下:
iqtree -s example.phy -m MPF -mset WAG,LG,JTT -mrate G,I,I+G -mfreq FU,F
指定模型参数设置格式:-m MODEL+FreqType+RateType,
MODEL:model name
+FreqType:(可选项)frequency type
+RateType:(可选项)rate heterogeneity type
替换模型MODEL包括:
- DNA模型:
JC/JC69, F81, K2P/K80, HKY/HKY85, TN/TrN/TN93, TNe,
K3P/K81, K81u, TPM2, TPM2u, TPM3, TPM3u, TIM, TIMe,
TIM2, TIM2e, TIM3, TIM3e, TVM, TVMe, SYM, GTR and 6-digit
- 蛋白质模型:
BLOSUM62, cpREV, Dayhoff, DCMut, FLU, HIVb, HIVw, JTT,
JTTDCMut, LG, mtART, mtMAM, mtREV, mtZOA, mtMet, mtVer,
mtInv, Poisson, PMB, rtREV, VT, WAG
+FreqType碱基使用偏好:Base frequencies 可选设置:
每个核苷酸位点上的替代是随机发生的,则A,T,C,G出现的频率应该大致相等。实际情况:DNA受到自然选择的压力,各个位点的碱基出现频率并不相等。
+RateType:rate heterogeneity across sites可选设置: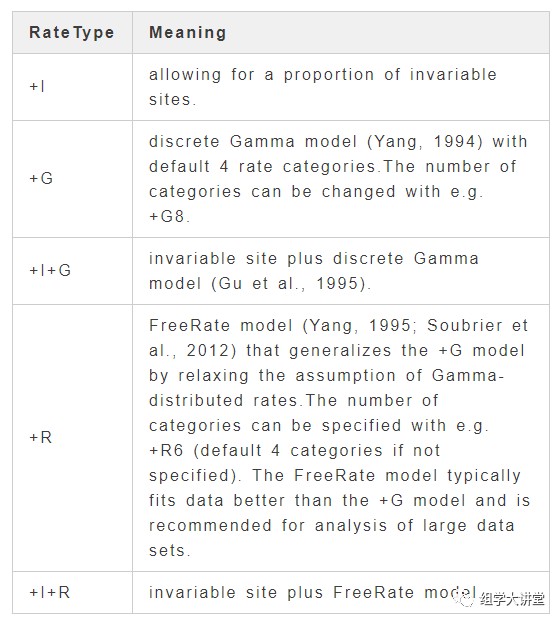
指定替换模型构建进化树命令举例:
iqtree -s example.phy -m TIM2+I+G
Bootstrap法评估分支支持度
真实的进化信息只有一个,而我们总是拿着有限的序列信息,希望去获得他。能否获得他,是一个问题。而我们使用的序列信息是否能真实且稳定地反应一个进化信息,那么是另外一个事情。bootstrap法常用的,尤其是ML法构建进化树上,分支可靠性检验方法。但是这个计算逻辑最大的问题在于,抽样重新跑,抽样再重新跑,不断重复,直到收敛或者是到指定的比如1000次。计算量大,耗时长。IQ-tree的作者团队在前述提出了一个快速的BS方法,最后整合到IQ-tree中。使用的方式是
iqtree -s example.phy -m TIM2+I+G -b 1000
超快(ultrafast bootstraping)
大概是IQTREE的精髓所在。顾名思义,ultrafast bootstrap approximation的特点就是超快。这里涉及到的细节,感兴趣的读者可以参见IQTREE的开发者写的几篇文章。作者认为,UFBoot is 10 to 40 times faster than RAxML rapid bootstrap and obtains less biased support values。
iqtree -s example.phy -m TIM2+I+G -B 1000
除ultrafast bootstrap之外,IQTREE还提供了以下检验树的拓扑结构可信度的方法。
- -alrt:SH-aLRT检验(4),没记错的话FastTree2使用的就是这个吧?
- -abayes:approximate Bayes test,由瑞士苏黎世应用科学教授Maria Anisimova提出(5)
- -lbp:fast local bootstrap probability method,由Adachi and Hasegawa提出(http://www.is.titech.ac.jp/~shimo/class/doc/csm96.pdf)
iqtree -s example.phy -m TIM2+I+G -B 1000 -alrt 1000
如果你指定了多个检验方法,那么其结果会在树里(.treefile)呈现出来,不同检验所得数值用斜线隔开,比如:((a,b)100/98:0.1,c:0.2)90/95最后,iqtree做群体遗传进化构建进化树推荐命令:
iqtree2 -s supergene.phy -st DNA -T 2 -mem 8G \
-m GTR -redo \
-B 1000 -bnni \
--prefix iqtree
3. RAxML 构建进化树
RAxML是最大似然法(maximum likelihood)建树的经典工具,其由来自德国海德堡理论科学研究所(Heidelberg Institute for Theoretical Studies)的Alexandros Stamatakis开发,最新已经更新了版本RAxML-NG,支持的替换模型更多,运行速度更快。
RAxML 建树原理
RAxML采用最大似然法建树,即将系统树的拓扑结构、分支长度及进化模型等的全部或者部分作为需要估计的参数,在给定的数据集及进化模型的基础上,用最大似然法的标准-似然值最大化来估计这些参数。首先,要选择进化模型,以简约树或者联接树为基础,采用似然法估计模型中各个参数。设置好参数后,以简约树或者联接树作为起始树,进行似然分析,最后用统计学方法从多个似然树中寻找最佳得分树。
RAxML-NG 使用
RAxML软件支持输入文件的格式可以是比对好的fasta格式或者phylip格式,例如DNA比对序列,核苷酸替代模型为GTR,rate heterogeneity设置为gamma分布,不做bootstraping,命令如下:
raxml-ng --msa supergene.phy --model GTR+G --prefix raxml_tree --threads 2 --seed 123
如果是建树和bootstrap一起做,可以加--all参数一步完成:
raxml-ng --msa supergene.phy --model GTR+G --prefix raxml_tree --threads 2 --seed 123
关于raxml-ng模型的设置与iq-tree类似,支持很多种个性化的模型设置:
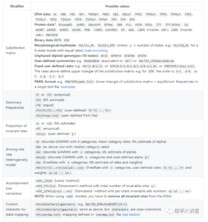
详细设置方法见:https://github.com/amkozlov/raxml-ng/wiki/Input-data#evolutionary-model
更多生物信息课程:https://study.omicsclass.com/index

- 发表于 2020-10-14 15:03
- 阅读 ( 15716 )
- 分类:遗传进化
