肠道微生物诊断慢性肾病-随机森林分析文献复现 IF=15分文章
肾脏的主要功能是保持体内平衡,包括酸碱平衡,水平衡,血压调节和葡萄糖稳态在内的功能需要肾脏进行精确的协调和调节。全球大约9%的人口正在遭受慢性肾脏疾病。近年来,研究表明肠道菌群和慢性肾病(CKD)相关,已有研究表明肠道菌群代谢的一些有毒副产品如氧化三甲胺(TMAO)甲酚硫酸酯(PCS),硫酸吲哚酚(IS)等会进入血液进而引起慢性肾病。

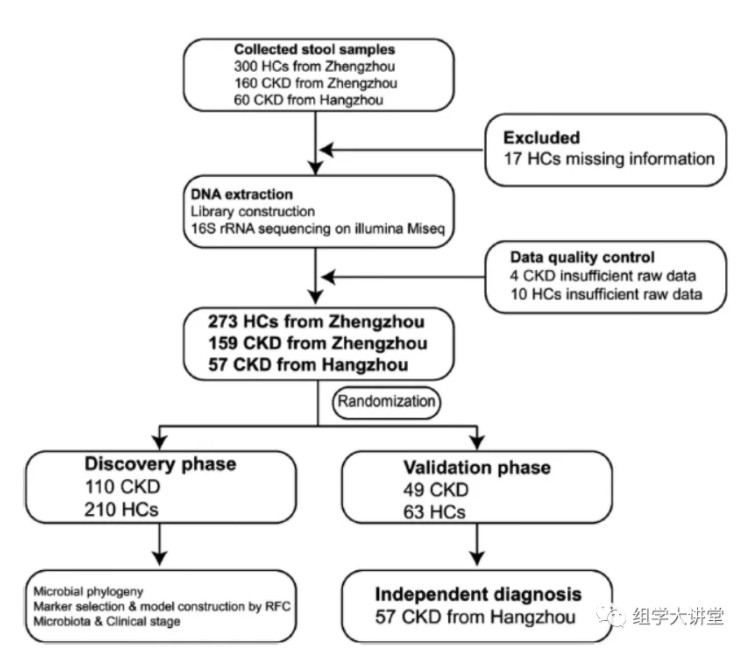
国内团队:浙江大学医学院附属第一医院的李兰娟院士团队与郑州大学附属第一医院的余祖江团队、刘章锁团队及吴歌。Advanced Science[IF:15.84] DOI: 10.1002/advs.202001936研究概述:该研究从中国郑州和杭州收集健康人与CKD患者的粪便样本共520份,通过微生物16S rRNA基因高通量测序,并用现在十分火热的机器学习的方法成功找到5个OTU可作为CKD的诊断标志物,可区分患者与对照AUC值达到0.9887。下面来解释一下作者是如何设计实验和分析的:实验设计:作者在郑州和杭州两地收集健康和CKD患者的粪便样本,通过数据质控最终在郑州收集到273(健康HCs)和159(CKD患者)样本,在杭州收集57份CKD样本。作者将这些样本分成三份分别作为随机森林分析的训练(discovery),测试(validation),独立验证预测(independet)数据集。
结果解读:
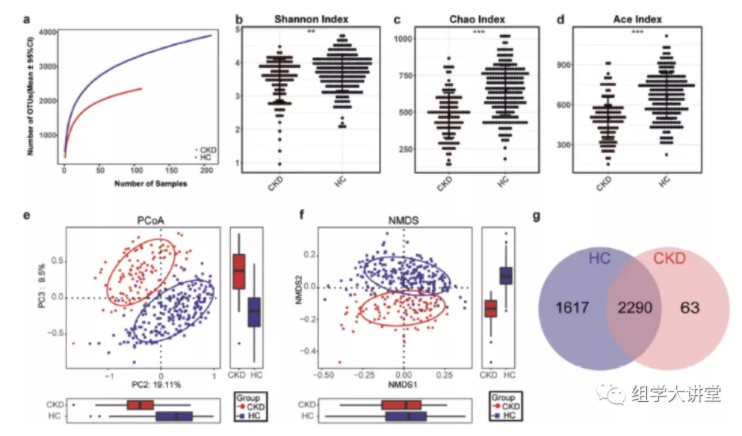
通过alpha和beta多样性分析发现,CKD患者的肠道菌群多样性显著降低,且CKD患者的肠道菌群结构与正常人差异显著;
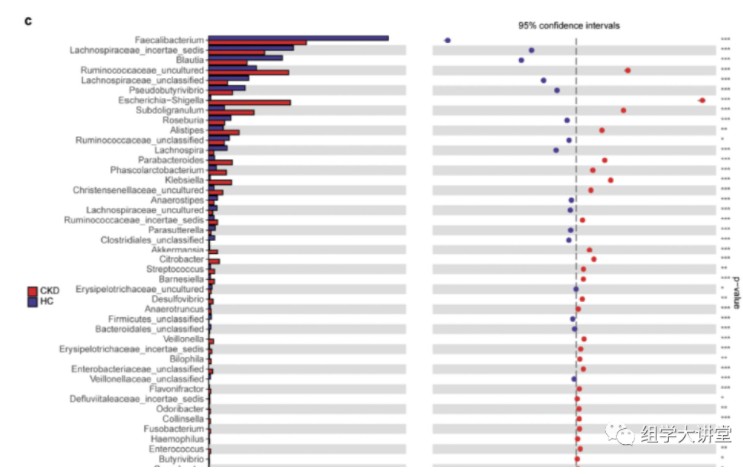
属水平物种差异比较发现:CKD患者Klebsiella属和Enterobacteriaceae科显著富集,而Blautia属和Roseburia属丰度降低。
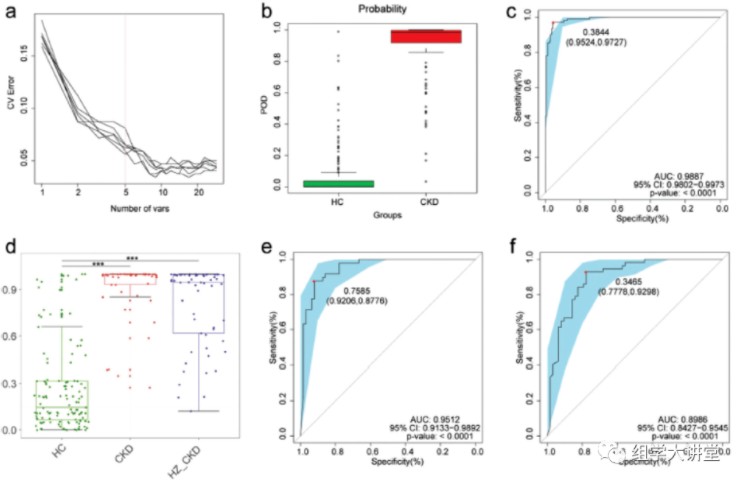
随机森林分析(不懂随机森林:点击):在训练数据集中构建模型用于对CKD患者和健康人进行分类,最终找到5个OTU作为biomarker可以准确预测CKD患者(下图a),预测指数POD在健康和患者中显著差异图b,ROC曲线分析预测准确性在训练数据集AUC 达0.9887 图c。该模型在测试数据集和独立验证数据集中都有很好的预测准确性(图d e f),说明有很好的普适性。
总结: 本文实验设计清晰,简单明了,纯生信分析能发到15分的文章,小编觉得文章的最大的亮点在于使用了当下热门的机器学习(随机森林)的方法,并且用到了三批数据反复验证,最终,找到与CKD预测相关的biomarker。这里小编下载了这篇文章中的部分数据,复现了一下文章中的随机森林分析结果,附带代码分享给大家:
1.随机森林分析注意事项
随机森林分析建议分组内样品至少30个以上,样品数太少影响准确性。随机森林分析,一般将数据划分为训练数据集(train data)和测试数据集(test or validate data),训练数据集用于构建预测模型,测试集用于查看模型的准确性。
如果想知道模型的普适性,应再增加独立验证数据(independent)集,以评估模型预测的普适性。
2.加载需要的包
#设置镜像,
local({r <- getOption("repos")
r["CRAN"] <- "http://mirrors.tuna.tsinghua.edu.cn/CRAN/"
options(repos=r)})
# 依赖包列表安装与加载:
package_list <- c("ggplot2","RColorBrewer","randomForest","caret", "pROC","dplyr","ggrepel")
# 判断R包加载是否成功来决定是否安装后再加载
for(p in package_list){
if(!suppressWarnings(suppressMessages(require(p, character.only = TRUE, quietly = TRUE, warn.conflicts = FALSE)))){
install.packages(p, warn.conflicts = FALSE)
suppressWarnings(suppressMessages(library(p, character.only = TRUE, quietly = TRUE, warn.conflicts = FALSE)))
}
}
3.随机森林之分类(classification)
3.1数据读取和建立模型
###############################################################################################
#读入物种丰度表格
otutable<-read.table("data/feature_table.txt",sep="\t",header = TRUE,check.names=FALSE,comment.char="",row.names= 1)
otutable[1:5,1:5]
## SRR10028875 SRR10028876 SRR10028877 SRR10028878 SRR10028879
## ASV_1 0 0 9 0 34
## ASV_2 0 0 13 0 2
## ASV_3 0 0 0 0 0
## ASV_4 0 0 1 0 0
## ASV_5 0 0 0 0 1
#读入fastmap信息
fastmap<-read.table("data/fastmap.txt",sep="\t",header = TRUE,check.names=FALSE,comment.char="",row.names= 1)
head(fastmap)
## SampleName tags source type phase group clinical_stage age
## SRR10028942 CKD_100 18587 zz CKD Discovery B 3 57
## SRR10028940 CKD_101 22183 zz CKD validation B 3 42
## SRR10028939 CKD_102 15117 zz CKD Discovery C 5 45
## SRR10028938 CKD_103 12276 zz CKD Discovery B 3 60
## SRR10028937 CKD_104 11363 zz CKD Discovery B 3 63
## SRR10028936 CKD_105 15489 zz CKD validation A 1 62
## gender BMI hypertension WBC RBC Hb PLT 24hUTP P BUN UA
## SRR10028942 male 24.71 1 6.5 4.7 146.0 137 3.43 1.01 9.0 380
## SRR10028940 male 25.10 1 6.7 5 156.0 172 0.39 1.11 6.1 314
## SRR10028939 female 24.17 0 6.7 2.99 89.0 164 1.10 2.81 33.9 687
## SRR10028938 male 23.68 0 66 4.89 132.0 249 1.00 0.74 5.3 263
## SRR10028937 male 23.18 1 8.71 5.04 149.3 242 0.78 1.09 11.3 512
## SRR10028936 female 22.78 1 7.1 4.92 150.0 233 1.13 1.33 3.6 306
## Scr eGFR ALB T_chol TG
## SRR10028942 189 33.191 46.1 6.03 2.35
## SRR10028940 131 57.000 50.4 4.09 1.53
## SRR10028939 578 9.348 36.1 3.98 0.80
## SRR10028938 124 54.000 42.0 4.90 2.62
## SRR10028937 196.6 35.413 39.8 4.09 2.67
## SRR10028936 55 96.840 45.1 4.74 2.19
#交叉筛选,使数据统一,以免不同文件ID不统一导致报错;以otutable为准筛选共同的ID
sampleID_inter=intersect(colnames(otutable),rownames(fastmap))
fastmap<-fastmap[sampleID_inter,]
otutable<-otutable[,sampleID_inter]
otutable.t<-t(otutable) #转置一下,使样品为行名,方便做随机森林分析
otutable.t<-as.data.frame(otutable.t) #转化成数据框格式
# 转化为因子,确保是分类数据;这里注意阳性组名称应放在levels 的第二个;
fastmap$type=factor(fastmap$type,order=T,levels=c("HC","CKD"))
fastmap$type
## [1] CKD CKD CKD CKD CKD CKD CKD CKD CKD CKD CKD CKD CKD CKD CKD CKD CKD CKD
## [19] CKD CKD CKD CKD CKD CKD CKD CKD CKD CKD CKD CKD CKD CKD CKD CKD CKD CKD
## [37] CKD CKD CKD CKD CKD CKD CKD CKD CKD CKD CKD CKD CKD CKD CKD CKD CKD CKD
## [55] CKD CKD CKD CKD CKD CKD CKD CKD CKD CKD CKD CKD CKD CKD CKD CKD CKD CKD
## [73] CKD CKD CKD CKD CKD CKD CKD CKD CKD CKD CKD CKD CKD CKD CKD CKD CKD CKD
## [91] CKD CKD CKD CKD CKD CKD CKD CKD CKD CKD CKD CKD CKD CKD CKD CKD CKD CKD
## [109] CKD CKD CKD CKD CKD CKD CKD CKD CKD CKD CKD CKD CKD CKD CKD CKD CKD CKD
## [127] CKD CKD CKD CKD CKD CKD CKD CKD CKD CKD CKD CKD CKD CKD CKD CKD CKD CKD
## [145] HC HC HC HC HC HC HC HC HC HC HC HC HC HC HC HC HC HC
## [163] HC HC HC HC HC HC HC HC HC HC HC HC HC HC HC HC HC HC
## [181] HC HC HC HC HC HC HC HC HC HC HC CKD CKD CKD HC CKD CKD CKD
## [199] CKD HC CKD CKD CKD CKD CKD CKD CKD HC CKD CKD CKD CKD CKD CKD CKD CKD
## [217] HC CKD CKD CKD CKD CKD CKD CKD CKD HC CKD CKD CKD CKD HC HC HC HC
## [235] HC HC HC HC HC HC HC HC HC HC HC HC HC HC HC HC HC HC
## [253] HC HC HC HC HC HC HC HC HC HC HC HC HC HC HC HC HC HC
## [271] HC HC HC HC HC HC HC HC HC HC HC HC HC HC HC HC HC HC
## [289] HC HC HC HC HC HC HC HC HC HC HC HC HC HC HC HC HC HC
## Levels: HC < CKD
#数据种添加需要预测的分组变量
otutable.t$type<-fastmap$type
fastmap_Discovery<-subset(fastmap,phase=="Discovery") #筛选Discovery的样本训练
fastmap_validation<-subset(fastmap,phase=="validation") #筛选validation的样本测试
trainset <- otutable.t[rownames(fastmap_Discovery),]
testset <- otutable.t[rownames(fastmap_validation),]
trainset[1:5,1:5]
## ASV_1 ASV_2 ASV_3 ASV_4 ASV_5
## SRR10028875 0 0 0 0 0
## SRR10028876 0 0 0 0 0
## SRR10028877 9 13 0 1 0
## SRR10028878 0 0 0 0 0
## SRR10028879 34 2 0 0 1
testset[1:5,1:5]
## ASV_1 ASV_2 ASV_3 ASV_4 ASV_5
## SRR10028888 6 19 0 0 0
## SRR10028890 2 124 0 0 0
## SRR10028894 74 39 0 0 0
## SRR10028900 2 5 0 0 0
## SRR10028901 3 283 0 0 0
set.seed(315) # 设置随机数种子,保证结果的可重复性,模型会收到随机数影响,可以多次尝试不同的随机数
rf.train <- randomForest(type~., # 分组作为目标变量
data=trainset, # 使用训练集构建随机森林
ntree = 1000, # 决策树的数量,默认的是1000
importance = TRUE, # 是否评估预测变量的重要性
proximity = TRUE) # 是否计算行之间的接近度
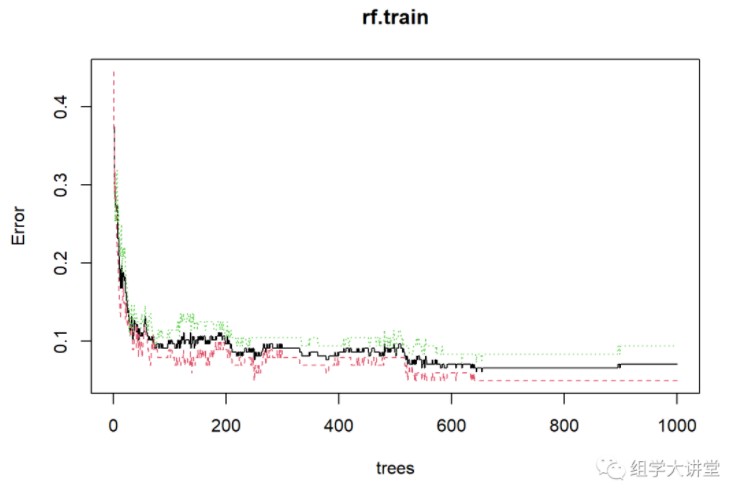
rf.train #初步看一下模型的预测准确性:准确率=1-OOB
##
## Call:
## randomForest(formula = type ~ ., data = trainset, ntree = 1000, importance = TRUE, proximity = TRUE)
## Type of random forest: classification
## Number of trees: 1000
## No. of variables tried at each split: 13
##
## OOB estimate of error rate: 7.11%
## Confusion matrix:
## HC CKD class.error
## HC 96 5 0.04950495
## CKD 9 87 0.09375000
plot(rf.train) #模型树的确定
3.2优化模型参数
3.2.1.选取重要的变量(物种)用于构建预测模型,通常30个以内
首先导出重要性,查看重要性结果:
imp= as.data.frame(rf.train$importance)
imp = imp[order(imp$MeanDecreaseAccuracy,decreasing = T),]
write.table(imp,file = "importance_feature.txt",quote = F,sep = '\t', row.names = T, col.names = T) #输出重要性
head(imp)
## HC CKD MeanDecreaseAccuracy MeanDecreaseGini
## ASV_23 0.037058297 0.048076271 0.042055566 9.244474
## ASV_104 0.023627631 0.032940311 0.028077878 6.840765
## ASV_32 0.011633950 0.017471974 0.014429591 3.991991
## ASV_129 0.016021674 0.008826792 0.012423272 3.580420
## ASV_28 0.012320317 0.008946866 0.010673991 2.561823
## ASV_122 0.006887519 0.011882535 0.009290654 2.667736
varImpPlot(rf.train) #重要性绘图
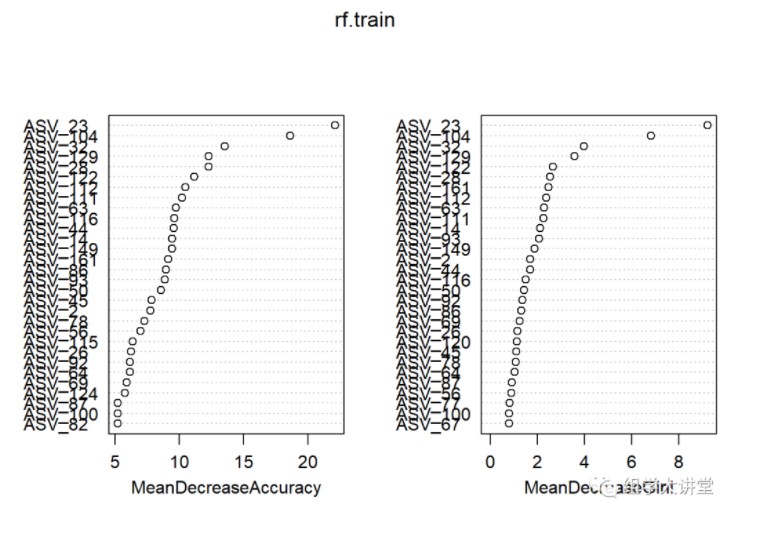
此处“Mean Decrease Accuracy”和“Mean Decrease Gini”为随机森林模型中的两个重要指标。其中,“mean decrease accuracy”表示随机森林预测准确性的降低程度,该值越大表示该变量的重要性越大;“mean decrease gini”计算每个变量对分类树每个节点上观测值的异质性的影响,从而比较变量的重要性。该值越大表示该变量的重要性越大
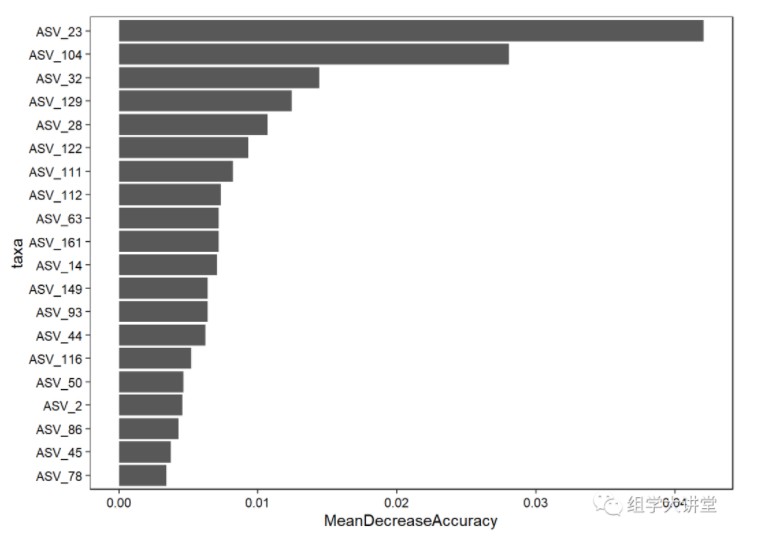
ggplot 美化贡献度绘图, 筛选前20个用于绘图
imp_sub=imp[1:20,]
imp_sub$taxa<-rownames(imp_sub)
# 添加因子保持顺序
imp_sub$taxa=factor(imp_sub$taxa,order=T,levels = rev(imp_sub$taxa))
p=ggplot(data = imp_sub, mapping = aes(x=taxa,y=MeanDecreaseAccuracy)) +
geom_bar(stat="identity")+coord_flip()+theme_bw()+
theme(panel.grid=element_blank(),
axis.text.x=element_text(colour="black"),
axis.text.y=element_text(colour="black"),
panel.border=element_rect(colour = "black"),
legend.key = element_blank(),plot.title = element_text(hjust = 0.5))
p
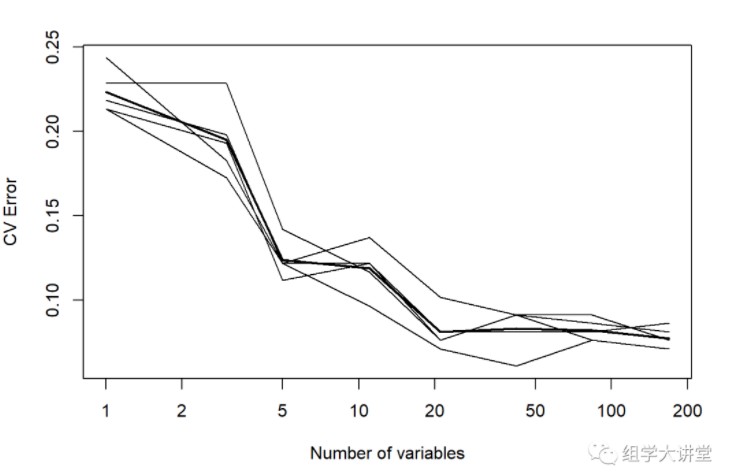
交叉验证确定最佳预测微生物数量:
英文:10-fold cross validation:
用来测试算法准确性。是常用的测试方法。将数据集分成十份,轮流将其中9份作为训练数据,1份作为测试数据,进行试验。每次试验都会得出相应的正确率(或差错率)。10次的结果的正确率(或差错率)的平均值作为对算法精度的估计,一般还需要进行多次10折交叉验证(例如10次10折交叉验证),再求其均值,作为对算法准确性的估计。
之所以选择将数据集分为10份,是因为通过利用大量数据集、使用不同学习技术进行的大量试验,表明10折是获得最好误差估计的恰当选择,而且也有一些理论根据可以证明这一点。但这并非最终诊断,争议仍然存在。而且似乎5折或者20折与10折所得出的结果也相差无几。
#这里采用10 fold,重复5次
set.seed(123)
result= replicate(5, rfcv(trainset[,-ncol(trainset)],trainset$type,cv.fold=10), simplify=FALSE)
error.cv= sapply(result, "[[", "error.cv")
matplot(result[[1]]$n.var, cbind(rowMeans(error.cv), error.cv), type="l",
lwd=c(2, rep(1, ncol(error.cv))), col=1, lty=1, log="x",
xlab="Number of variables", ylab="CV Error")
#输出交叉验证结果,方便后续美化出图,
cv.result=cbind(result[[1]]$n.var, rowMeans(error.cv), error.cv)
write.table(cv.result,file ="rfcv_result.txt",quote = F,sep = '\t', row.names = T, col.names = T)
cv.result
## [,1] [,2] [,3] [,4] [,5] [,6] [,7]
## 169 169 0.07715736 0.08121827 0.08629442 0.07106599 0.07106599 0.07614213
## 84 84 0.08223350 0.08629442 0.08121827 0.07614213 0.07614213 0.09137056
## 42 42 0.08324873 0.09137056 0.08121827 0.09137056 0.06091371 0.09137056
## 21 21 0.08121827 0.07614213 0.08121827 0.07614213 0.07106599 0.10152284
## 11 11 0.11878173 0.12182741 0.12182741 0.11675127 0.09644670 0.13705584
## 5 5 0.12385787 0.11167513 0.12182741 0.14213198 0.12182741 0.12182741
## 3 3 0.19492386 0.19289340 0.19796954 0.22842640 0.17258883 0.18274112
## 1 1 0.22335025 0.21319797 0.21827411 0.22842640 0.21319797 0.24365482
第一列为 “Number_of_variables” 第二列:“mean_error_rate” 其他列为重复的error_rate3.2.2.优化参数:mtry,也就是每棵树用到的变量个数更多参数优化校准方法:https://rpubs.com/phamdinhkhanh/389752 或者 https://topepo.github.io/caret/model-training-and-tuning.html
#确定用于模型构建的数量后,
#再优化模型选择最佳mtry,也就是每课树中的预测变量数量
#这里选择前10个重要的变量,并优化选择最佳mtry
rf.formula=as.formula(paste0("type ~",paste(row.names(imp)[1:10],collapse="+")))
rf.formula
## type ~ ASV_23 + ASV_104 + ASV_32 + ASV_129 + ASV_28 + ASV_122 +
## ASV_111 + ASV_112 + ASV_63 + ASV_161
#校准模型mtry
train.res=train(rf.formula,
data = trainset, # Use the train data frame as the training data
method = 'rf',# Use the 'random forest' algorithm
trControl = trainControl(method='cv',
number=10,
repeats=5,
search='grid')) # Use 10 folds for cross-validation 重复5次
## Warning: `repeats` has no meaning for this resampling method.
train.res
## Random Forest
##
## 197 samples
## 10 predictor
## 2 classes: 'HC', 'CKD'
##
## No pre-processing
## Resampling: Cross-Validated (10 fold)
## Summary of sample sizes: 177, 178, 177, 177, 177, 178, ...
## Resampling results across tuning parameters:
##
## mtry Accuracy Kappa
## 2 0.8813158 0.7631431
## 6 0.8968421 0.7941472
## 10 0.8968421 0.7941472
##
## Accuracy was used to select the optimal model using the largest value.
## The final value used for the model was mtry = 6.
#选择最佳:mtry
model <- randomForest(rf.formula, # 新建的模型
data=trainset, # 使用训练集构建随机森林
ntree = 500, # 决策树的数量,默认的是500
mtry = 2, # 每个分组中随机的变量数,默认是变量数开根
importance = TRUE, # 是否评估预测变量的重要性
proximity = TRUE) # 是否计算行之间的接近度
model
##
## Call:
## randomForest(formula = rf.formula, data = trainset, ntree = 500, mtry = 2, importance = TRUE, proximity = TRUE)
## Type of random forest: classification
## Number of trees: 500
## No. of variables tried at each split: 2
##
## OOB estimate of error rate: 9.14%
## Confusion matrix:
## HC CKD class.error
## HC 93 8 0.07920792
## CKD 10 86 0.10416667
可以看到模型在训练数据集中的准确率很高:1-oob
用测试数据集测试预测准确性
pred1 <- predict(model, newdata = testset,type="response")
#预测结果
confusionMatrix(pred1, testset$type)
## Confusion Matrix and Statistics
##
## Reference
## Prediction HC CKD
## HC 25 6
## CKD 2 42
##
## Accuracy : 0.8933
## 95% CI : (0.8006, 0.9528)
## No Information Rate : 0.64
## P-Value [Acc > NIR] : 6.162e-07
##
## Kappa : 0.7758
##
## Mcnemar's Test P-Value : 0.2888
##
## Sensitivity : 0.9259
## Specificity : 0.8750
## Pos Pred Value : 0.8065
## Neg Pred Value : 0.9545
## Prevalence : 0.3600
## Detection Rate : 0.3333
## Detection Prevalence : 0.4133
## Balanced Accuracy : 0.9005
##
## 'Positive' Class : HC
##
可以看到准确性:Accuracy :
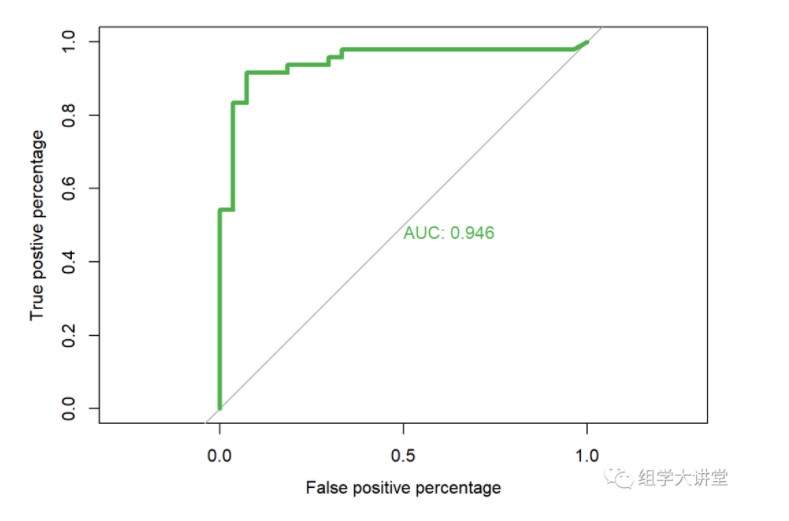
3.3 模型准确性评估 ROC曲线,并计算AUC值
pred1 <- predict(model, newdata = testset,type="vote")
roc.info<-roc(testset$type,
pred1[,1], #提取随机森林模型中对应的预测指标
plot=TRUE,
legacy.axes=TRUE,
percent=FALSE,
xlab="False positive percentage",
ylab="True postive percentage",
col="#4daf4a",
lwd=4,
print.auc=TRUE)
## Setting levels: control = HC, case = CKD
## Setting direction: controls > cases
课程推荐:微生物扩增子分析课程实操 微生物16S/ITS/18S分析原理及结果解读
延伸阅读:
1. 微生物测序原理2. 肠道“君”与人类二三事(科普)3. 什么是OTU4. alpha多样性5. GraPhlAn树状图6. OTU网络图MENA7. beta多样性解读
8. 一文读懂微生物多样性中的各种排序分析方法异同点9. 微生物组间差异分析之LEfSe分析10. 用测序研究微生物多样性,您选对区了吗?11.微生物多样性分析docker镜像使用12.生成OTU方法如何选择,去噪还是聚类?13.微生物多样性组间差异分析—STAMP使用!
14.16S rDNA你知道多少?
- 发表于 2020-12-01 15:59
- 阅读 ( 8318 )
- 分类:宏基因组
