STAR比对软件的使用
1. 下载安装(linux)
wget -c https://github.com/alexdobin/STAR/archive/2.7.3a.tar.gz
tar -xvzf 2.7.3a.tar.gz
cd STAR/source
make STAR
2. STAR的使用
分为两部分:建索引和序列比对
(1)建索引
STAR --runThreadN 6 \
--runMode genomeGenerate \
--genomeDir GRCm38 \
--genomeFastaFiles GRCm38.p5.genome.fa \
--sjdbGTFfile gencode.vM13.annotation.gtf \
--sjdbOverhang 125
参数说明:
--runThreadN:设置线程数,线程数越大越占内存
--runMode:运行模式,默认为比对,第一步一定要设置。
--genomeDir:生成的索引文件输出目录
--genomeFastaFiles:基因组fasta文件
--sjdbGTFfile:GTF注释文件
--sjdbOverhang:reads长度减1
构建成功后会在指定的输出目录下生成如下文件:
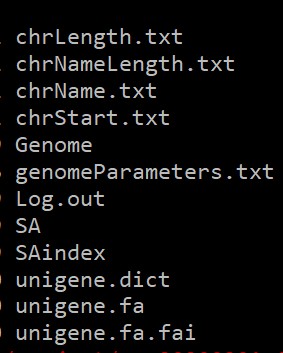 (2)序列比对
(2)序列比对
STAR--genomeDir SNP_Analysis/genomeDir \
--readFilesInsample1.clean.fq.gz sample2.clean.fq.gz \
--runThreadN4 \
--sjdbOverhang 125 \
--readFilesCommand zcat \
--outSAMtype BAM SortedByCoordinate \
--twopassMode Basic
参数说明:
--runThreadN:设置线程数,线程数越大越占内存
--genomeDir:生成的索引文件输出目录
--readFilesIn:原始测序文件
--sjdbOverhang:值为测序read的长度减1,是在注释可变剪切序列的时使用的最大长度值
--readFilesCommand:读文件的命令,说明gz结尾的压缩文件
--outSAMtype:输出文件格式(默认输出排序的BAM文件)
--twopassMode:(None/Basic)为了发现更加灵敏的new junction,建议使用2-pass mode,其能增加检测到的new junction数目,使得更多的splices reads能mapping到new junction
运行过程:
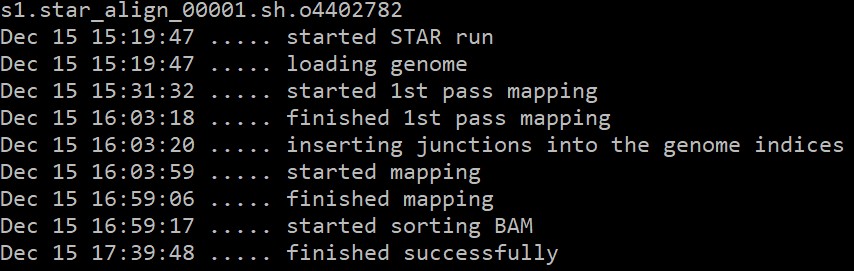
输出文件:
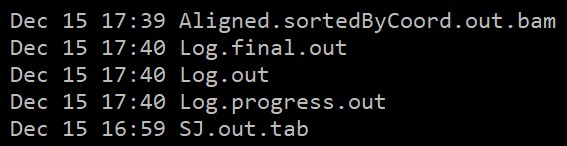
参考文章:https://pubmed.ncbi.nlm.nih.gov/23104886/
此外,我们在网易云课堂上有各种教学视频,有兴趣可以了解一下:
1. 文章越来越难发?是你没发现新思路,基因家族分析发2-4分文章简单快速,学习链接:基因家族分析实操课程
2. 转录组数据理解不深入?图表看不懂?点击链接学习深入解读数据结果文件,学习链接:转录组(有参)结果解读;转录组(无参)结果解读
3. 转录组数据深入挖掘技能-WGCNA,提升你的文章档次,学习链接:WGCNA-加权基因共表达网络分析
4. 转录组数据怎么挖掘?学习链接:转录组标准分析后的数据挖掘
6. 更多学习内容:linux、perl、R语言画图,更多免费课程请点击以下链接:
http://m.study.163.com/provider/400000000234009/index.htm?share=1&shareId=1433306807
- 发表于 2020-12-17 10:54
- 阅读 ( 7196 )
- 分类:软件工具
