免疫疫微环境(TIME)
肿瘤免疫微环境(Tumor immune microenvironment)是指肿瘤细胞存在的周围微环境,包括周围的血管、免疫细胞、成纤维细胞、骨髓源性炎性细胞、各种信号分子和细胞外基质。肿瘤和周围环境密切相关,不断进行交互作用,肿瘤可以通过释放细胞信号分子影响其微环境,促进肿瘤的血管生成和诱导免疫耐受,而微环境中的免疫细胞可影响癌细胞增长和发育。
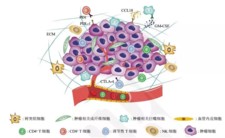
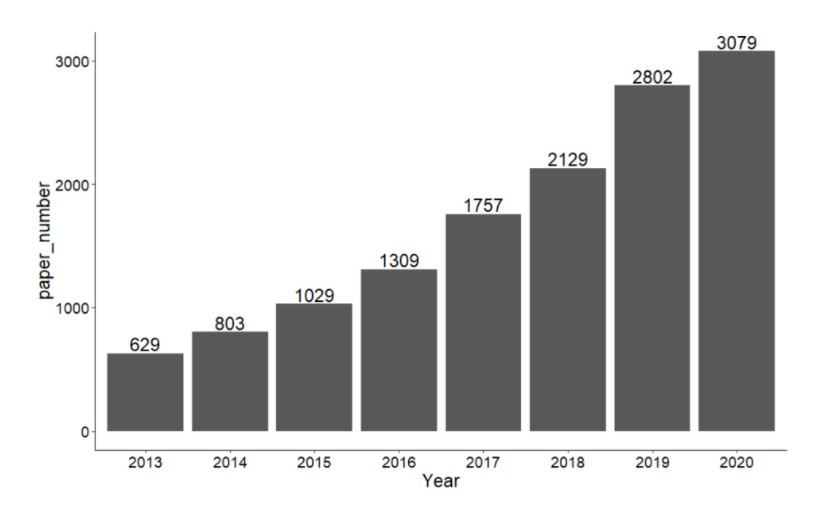
对于免疫微环境有一个流行的学说——“种子与土壤”学说,肿瘤的发生发展是肿瘤细胞与其微环境相互影响、共同进化的结果。肿瘤微环境由不同种类的间质细胞和炎性介质以及细胞外基质(ECM)组成。其中肿瘤相关成纤维细胞是最主要的间质细胞;血管内皮细胞介导的血管新生为肿瘤生长和转移提供必需的营养;免疫浸润包括树突状细胞、巨噬细胞、NK细胞及不同亚型的T细胞等等。肿瘤相关巨噬细胞与肿瘤细胞可以通过分泌特殊的细胞因子形成正反馈循环促进肿瘤恶性表型的形成和维持。肿瘤微环境在肿瘤恶性进展、免疫逃逸和治疗抵抗中发挥重要作用,我们统计了近8年(截至2020.11)pubmed 收录的关于肿瘤免疫浸润的文章数目,可见一直呈增长趋势,热度未减。
02
TIME的免疫特点和相关机制
肿瘤细胞和基质成分之间相互作用,形成了功能复杂的TME。肿瘤相关成纤维细胞(CAFs)主要分布于血管周围或肿瘤外周纤维间质内,分泌细胞因子、ECM成分及相关酶分子。TME中有多种免疫浸润细胞,其中CD8+或细胞毒性T淋巴细胞(CTL)发挥肿瘤杀伤功能,而调节型T细胞(Treg)减弱T细胞活性,促进TME免疫抑制。一般M1型巨噬细胞发挥促炎和抗瘤作用,但TME中的肿瘤相关巨噬细胞(TAM)为M2型,通过分泌Th2细胞因子促进血管生成和肿瘤侵袭。我们所熟知的NK细胞会释放颗粒酶和穿孔素杀伤靶细胞,但在TME中富集的TGF-β会抑制其杀伤活性。而树突状细胞(DC)也会受到TME中的缺氧和炎症影响消弱其抗原呈递活性。基质细胞类型和富集程度决定了TME特性,进一步影响着肿瘤进展和免疫应答情况。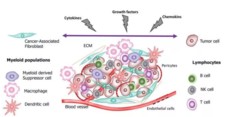
TME的异质性使得个体间肿瘤进展存在很大差异;肿瘤的免疫微环境一般分为豁免型和炎症型。炎症型肿瘤微环境中富集有活化的T细胞和髓系细胞,并由趋化因子、I型干扰素信号表达。相反“冷肿瘤”,既是免疫豁免型TME中,仅存在少量免疫细胞或抑制性亚群,如Treg、MDSC和TAM,而效应型免疫细胞无法有效浸润至肿瘤微环境。仅分布在外周基质,难以发挥抑癌作用。
03
检测免疫浸润的工具
肿瘤浸润情况对癌症治疗效果和患者预后有很大的影响,了解肿瘤微环境中免疫细胞的组成有助于揭示肿瘤异质性。利用RNA-seq技术进行基因表达谱分析可以表征肿瘤相关基因的表达谱,已广泛用于在许多癌症类型的研究。RNA-seq虽能提供基因表达信息,但并不能直接表征出免疫浸润情况,需要一定的算法进一步评估。
3.1
强大的TIMER2.0
TIMER2.0 (http://timer.cistrome.org/) 是TIMER软件的更新版本,作为一个可交互式web工具,能够全面、灵活的分析肿瘤免疫浸润免疫细胞并可视化。另外相关文章也是发表在了Nucleic Acids Research(IF=11.1)杂志上。

现有用于免疫浸润评估的算法可分为两大类:基于标记基因的肿瘤浸润免疫细胞量化方法和基于表达特征对细胞混合物进行反褶积的肿瘤浸润免疫细胞量化方法。基于特征基因集的方法是通过使用组织样本中这些特征基因集的表达量,对基因集进行富集分析或将其汇总到丰度评分中来独立推断每种细胞类型的得分;其中代表的软件有TIminer、xCell、MCP-counter。反卷积方法推断出数学方程式,该数学方程式是将组织样本的基因表达建模为种群混合物中细胞表达谱的加权总和。这两种互补的算法在估计不同肿瘤中特定的免疫细胞类型方面表现出不同的性能优势;相关软件有CIBERSORT、TIMER、EPIC、quanTIseq。另外还有一些评定免疫得分的软件如ESTIMATE,以及基于GSVA算法的ssGSEA。
TIMER2.0版本主要包含Immune, Exploration, Estimate三大模块,可以手动输入RNA-seq的基因TPM标准化值,TIMER2.0综合六个软件的优势(TIMER, xCell, MCP-counter, CIBERSORT, EPIC和quanTIseq)给出120个免疫微环境相关细胞的得分;另外该软件还将TCGA数据库中的所有样本用这六个软件计算出了免疫得分,另外还提供了很多可视化的线上工具。大家感兴趣的话,可以看看这篇论文,去线上平台尝试一下。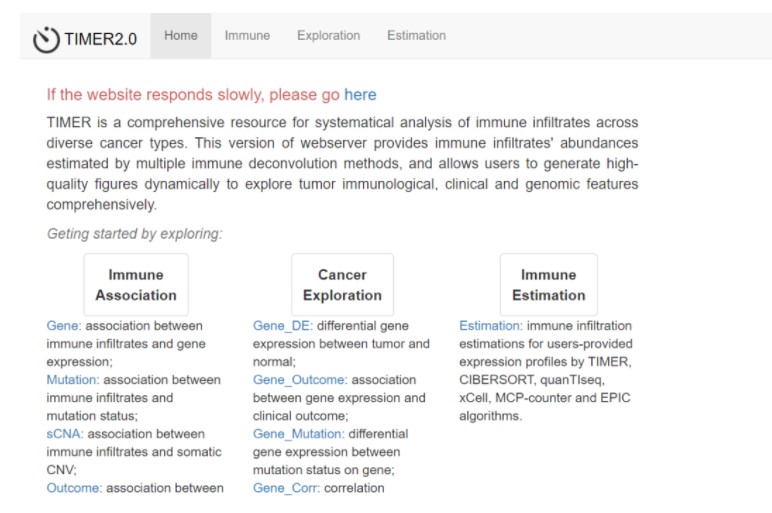
3.2
xCELL
xCell (http://xcell.ucsf.edu/) 是2017年发表在Genome Biology杂志上公布的基于ssGSEA的方法工具,该方法可估计64种免疫细胞类型的丰度分数,包括适应性和先天免疫细胞,造血祖细胞,上皮细胞和细胞外基质细胞。

xCell基于从不同项目和研究的大规模表达数据中提取的489个基因集(FANTOM5, ENCODE, Blueprint, IRIS, Human Primary Cell Atlas (HPCA)和Novershtern等人的文章)。对于每种细胞类型,通过四个主要步骤计算xCell丰度得分:(1)使用R包GSVA对489个基因集单独进行ssGSEA;(2)在属于一种细胞类型的所有基因集的ES进行平均;(3)将特定平台的ES转换为丰度分数;(4)使用类似于用于流式细胞术数据分析的spillover方法校正紧密相关的细胞类型之间的相关性。尽管最终的xCell丰度分数不能直接解释为细胞分数,但它们与真实的细胞比例具有很高的相关性。
另xCell还提供R包工具:####xCell包安装
options(stringsAsFactors = F)
Sys.setlocale("LC_ALL","English")
devtools::install_github('dviraran/xCell')
library(xCell)
##利用xCell分析RNA-seq数据
result <- xCellAnalysis(exprSet,rnaseq=T)
save(result,file = 'Xcell_result')
3.3
MCP-counter
MCP-counter (http://github.com/ebecht/MCPcounter) 是2016年发表在Genome Biology杂志上的一种基于标记基因集来量化肿瘤浸润免疫细胞、成纤维细胞和上皮细胞的方法。对于每种细胞类型和样品,将丰度得分计算为细胞类型特异性基因表达值的几何平均值。
由于分数以任意单位表示,因此不能直接将其解释为细胞分数,也不能在细胞类型之间进行比较。但是,使用定量验证显示估计分数与真实细胞分数之间具有高度相关性,从而证明了MCP-counter在样品间比较中的价值。为了证明这些估计的预后价值,MCP-counter已用于量化32个非血液学肿瘤中19000多个样本中的免疫细胞和非免疫细胞。
MCP-counter的R包脚本:
library(devtools)
install_github('ebecht/MCPcounter',ref='master',subdir = 'Source')
library(MCPcounter)
input <- read.table("RNA-seq_data ",sep = "\t",header = TRUE,row.names = 1)
#小编在运行时遇到了github连接的错误;设置C:\Windows\System32\drivers\etc\hosts文件;在文件最后面加:“199.232.68.133 raw.githubusercontent.com”
MCPcounter_estimate <- MCPcounter.estimate(input,featuresType = "HUGO_symbols")
heatmap(as.matrix(ExampleEstimates),col=colorRampPalette(c("blue","white","red"))(100))
3.4
CIBERSORT
CIBERSORT算法 (https://cibersort.stanford.edu/index.php) 考虑了由microarray数据构建的特征矩阵,22种免疫细胞表型的表达特征,包括不同的细胞类型和功能状态的免疫细胞。CIBERSORT使用ν-SVR估计细胞得分。通过对血液和淋巴结活检细胞混合物的microarray数据进行验证,CIBERSORT被证明在在9个免疫细胞亚群和3个免疫细胞亚群的同时反褶积方面具有较高的准确性。通过对四种恶性免疫细胞类型的模拟混合物进行测试,它还证明了对不同程度的噪音和未知的肿瘤具有稳定性。
线上工具直接提交RNA-seq的表达量值就可以得到免疫微环境的22种细胞的得分。

3.5
ESTIMATE
Estimate是2013年发表在Nature communication杂志上的一种推断肿瘤微环境中免疫得分,基质得分及免疫微环境得分的工具。主要是利用能够表征基质和免疫细胞的基因集的表达信号进行估算。
ESTIMATE方法估计是基于TCGA数据库中的各种平台检测的样本表达量数据得到的10412个基因集,经过过滤最终确定基质信号基因集的141个基因和免疫信号的141个基因,通过这两个基因集的表达量通过ssGSEA算法最终确定Stromal score和Immune score,这两个得分相加即可得到Estimate score;这三个得分分别表征着免疫微环境中基质细胞的占比、免疫细胞的占比和肿瘤纯度。
ESTIMATE的R包脚本:
library(utils)
rforge <- "http://r-forge.r-project.org"
install.packages("estimate", repos=rforge, dependencies=TRUE)
library(estimate)
help(package="estimate")
SCLC_Expr = "C:/Users/lxz/Desktop /sclc_ucologne_2015/data_RNAseq_SCLC_uni_sort_TPM"
filterCommonGenes(input.f=SCLC_Expr,output.f = "SCLC_10412genes.gct",id = "GeneSymbol")
estimateScore(input.ds = "SCLC_10412genes.gct",
output.ds="SCLC_estimate_score.gct",
platform="illumina")
##此步点图是基于Affymetrix平台的测序数据
#plotPurity(scores="SCLC_estimate_score.gct", samples="all_samples",platform="Affymetrix")
sclc_scores=read.table("SCLC_estimate_score.gct",skip = 2,header = T)
rownames(sclc_scores)=sclc_scores[,1]
sclc_scores=t(sclc_scores[,3:ncol(sclc_scores)])
write.table(sclc_scores,"Immunity_score",row.names = T,sep = "\t",quote = F)
好啦!关于肿瘤免疫微环境的介绍就到此为止啦,感兴趣的同学可以利用手头的RNA-seq数据赶紧操练起来啦!先预祝您能得到一个好结果!
- 发表于 2021-04-02 16:34
- 阅读 ( 22467 )
- 分类:TCGA
