ConsensusClusterPlus 包对基因表达数据进行一致性聚类
Consensus Clustering(一致性聚类)是一种无监督聚类方法,是一种常见的癌症亚型分类研究方法,可根据不同组学数据集将样本区分成几个亚型,从而发现新的疾病亚型或者对不同亚型进行比较分析。这类文章一般会对基因表达量(芯片数据或者RNA-seq数据)或甲基化等数据进行聚类分析,选出最优聚类数;对聚出的类组进行差异化表达分析得到DEGs,差异表达基因做GO、pathway,PPI等一系列分析,在分析一下与生存的关系、免疫细胞丰度的区别,等等。找出了分之间免疫细胞有区别、生存有区别,一篇揭示XX癌免疫应答异质性的文章就来了。
Consensus Clustering 的基本原理假设:从原数据集不同的子类中提取出的样本构成一个新的数据集,并且从同一个子类中有不同的样本被提取出来,那么在新数据集上聚类分析之后的结果,无论是聚类的数目还是类内样本都应该和原数据集相差不大。因此所得到的聚类相对于抽样变异越稳定,我们越可以相信这一样的聚类代表了一个真实的子类结构。重采样的方法可以打乱原始数据集,这样对每一次重采样的样本进行聚类分析然后再综合评估多次聚类分析的结果给出一致性(Consensus)的评估。总结,一致聚类通过基于重采样的方法来验证聚类合理性,其主要目的是评估聚类的稳定性,可用于确定最佳的聚类数目K。。
相比其他聚类方法一致性聚类的优势:
不能提供“客观的”分类数目的标准和分类边界,例如Hierarchical Clustering。
需要预先给定一个分类的数目,且没有统一的标准去比较不同分类数目下分类的结果,例如K-means Clustering。
聚类结果的合理性和可靠性无法验证。
R实现一致性聚类
ConsensusClusterPlus则将Consensus Clustering在 R 中实现了。
#安装包
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("ConsensusClusterPlus")
主要方法:
(1)ConsensusClusterPlus 方法用于一致性聚类,
ConsensusClusterPlus(d=NULL, maxK = 3, reps=10, pItem=0.8,
pFeature=1, clusterAlg="hc",
title="untitled_consensus_cluster",
innerLinkage="average",
finalLinkage="average",
distance="pearson", ml=NULL,
tmyPal=NULL,seed=NULL,
plot=NULL,writeTable=FALSE,weightsItem=NULL,
weightsFeature=NULL,verbose=F,corUse="everything")
常用参数:
d | 提供的需要聚类的数据矩阵,其中列是样本,行是features,可以是基因表达矩阵。 |
maxK | 聚类结果中分类的最大数目,必须是整数。 |
reps | 重抽样的次数 |
pItem | 样品的抽样比例,如 pItem=0.8 表示采用重抽样方案对样本的80%抽样,经过多次采样,找到稳定可靠的亚组分类。 |
pFeature | Feature的抽样比例 |
clusterAlg | 使用的聚类算法,“hc”用于层次聚类,“pam”用于PAM(Partioning Around Medoids)算法,“km”用于K-Means算法,也可以自定义函数。 |
title | 设置生成的文件的路径 |
| distance | 计算距离的方法,有pearson、spearman、euclidean、binary、maximum、canberra、minkowski。 |
tmyPal | 可以指定一致性矩阵使用的颜色,默认使用白-蓝色 |
seed | 设置随机种子。 |
plot | 不设置时图片结果仅输出到屏幕,也可以设置输出为'pdf', 'png', 'pngBMP' 。 |
writeTable | 若为TRUE,则将一致性矩阵、ICL、log输出到CSV文件 |
weightsItem | 样品抽样时的权重 |
weightsFeature | Feature抽样时的权重 |
verbose | 若为TRUE,可输出进度信息在屏幕上 |
corUse | 设置如何处理缺失值: all.obs:假设不存在缺失数据——遇到缺失数据时将报错 everything:遇到缺失数据时,相关系数的计算结果将被设为missing complete.obs:行删除 pairwise.complete.obs:成对删除,pairwisedeletion |
(2)calcICL函数:
用法:
calcICL(res,title="untitled_consensus_cluster",plot=NULL,writeTable=FALSE)
参数:
res | consensusClusterPlus的结果 |
title | 设置生成的文件的路径 |
plot | 不设置时图片结果仅输出到屏幕,也可以设置输出为'pdf', 'png', 'pngBMP' 。 |
writeTable | 若为TRUE,则将一致性矩阵、ICL、log输出到CSV文件 |
数据分析
首先收集用于聚类分析的数据,比如 mRNA 表达微阵列或免疫组织化学染色强度的实验结果数据。输入数据的格式应为矩阵。下面以 ALL 基因表达数据为例进行操作。
##使用ALL示例数据library(ALL)data(ALL)d=exprs(ALL)d[1:5,1:5] #共128个样品,12625个探针数据
# 01005 01010 03002 04006 04007 # 1000_at 7.597323 7.479445 7.567593 7.384684 7.905312 # 1001_at 5.046194 4.932537 4.799294 4.922627 4.844565 # 1002_f_at 3.900466 4.208155 3.886169 4.206798 3.416923 # 1003_s_at 5.903856 6.169024 5.860459 6.116890 5.687997 # 1004_at 5.925260 5.912780 5.893209 6.170245 5.615210
##对这个芯片表达数据进行简单的normalization,取在各个样品差异很大的那些gene或者探针的数据来进行聚类分析 mads=apply(d,1,mad) #计算每个基因的标准差 d=d[rev(order(mads))[1:5000],]
#sweep函数减去中位数进行标准化 d = sweep(d,1, apply(d,1,median,na.rm=T)) #也可以对这个d矩阵用DESeq的normalization 进行归一化,取决于具体情况
聚类分析
title="F:/ConsensusClusterPlus" #设置图片输出路径 results <- ConsensusClusterPlus(dataset, maxK = 6, reps = 50, pItem = 0.8, pFeature = 0.8, clusterAlg = "hc", seed=100, distance = "pearson", title = title, plot = "png")
#结果将会输出k从2-6各个情况下的分型情况,聚类的方法用的是 hc ,抽样比例为0.8,最后输出png图#这里设置的maxK=6、reps=50,但是实际上需要更高的reps(如1000)和更高的maxK(如20)
结果展示与说明;
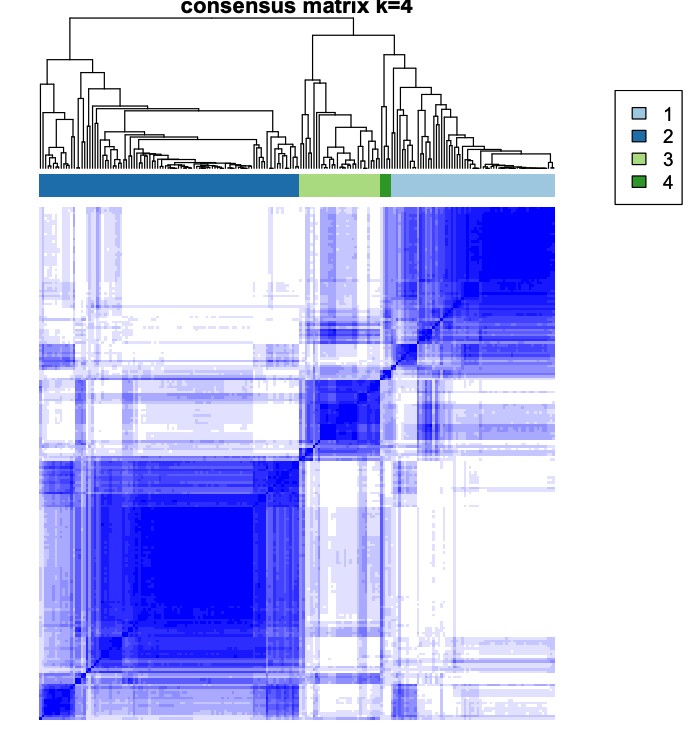
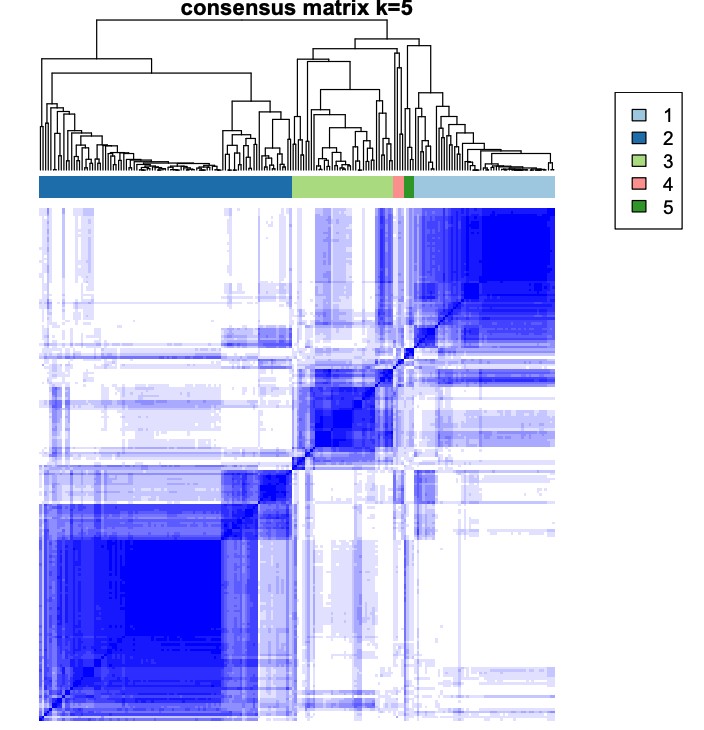
(1)k = 2, 3, 4, 5, 6 时的矩阵热图:矩阵的行和列表示的都是样本,一致性矩阵的值按从0(不可能聚类在一起)到1(总是聚类在一起)用白色到深蓝色表示,一致性矩阵按照一致性分类(热图上方的树状图)来排列。树状图和热图之间的长条即分出来的类别。注意第一张为图例;


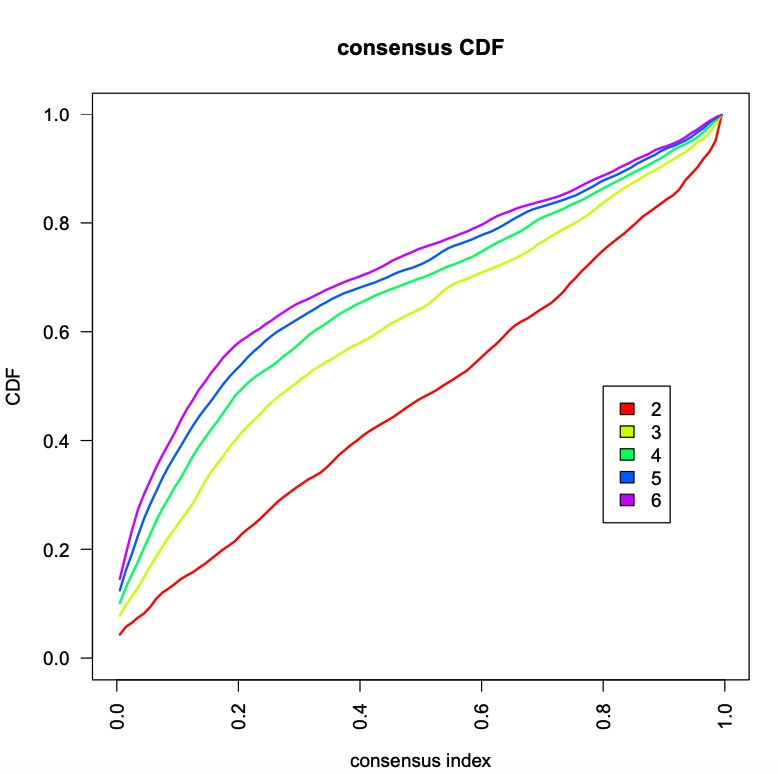
(2)一致性累积分布函数(CDF)图:此图展示了k取不同数值时的累积分布函数,用于判断当k取何值时,CDF达到一个近似最大值,此时的聚类分析结果最可靠。即考虑CDF下降坡度小的k值。
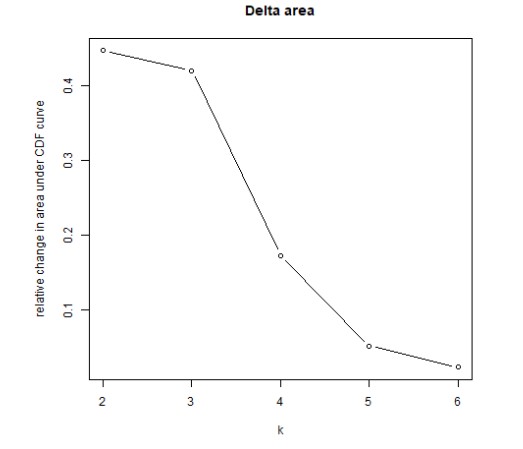
(3)Delta Area Plot:此图展示的是 k 和 k-1 相比CDF曲线下面积的相对变化。当k=2时,因为没有k=1,所以第一个点表示的是k=2时CDF曲线下总面积,而非面积的相对变化值。当k=6时,曲线下面积仅小幅增长,故5为合适的k值。
分类树及结果:
results[[2]][["consensusTree"]] # Call:# hclust(d = as.dist(1 - fm), method = finalLinkage) # # Cluster method : average # Number of objects: 128 results[[2]][["consensusClass"]][1:5] # 01005 01010 03002 04006 04007 # 1 1 1 1 1
计算聚类一致性 (cluster-consensus) 和样品一致性 (item-consensus)
icl <- calcICL(results, title = title,plot = "png")
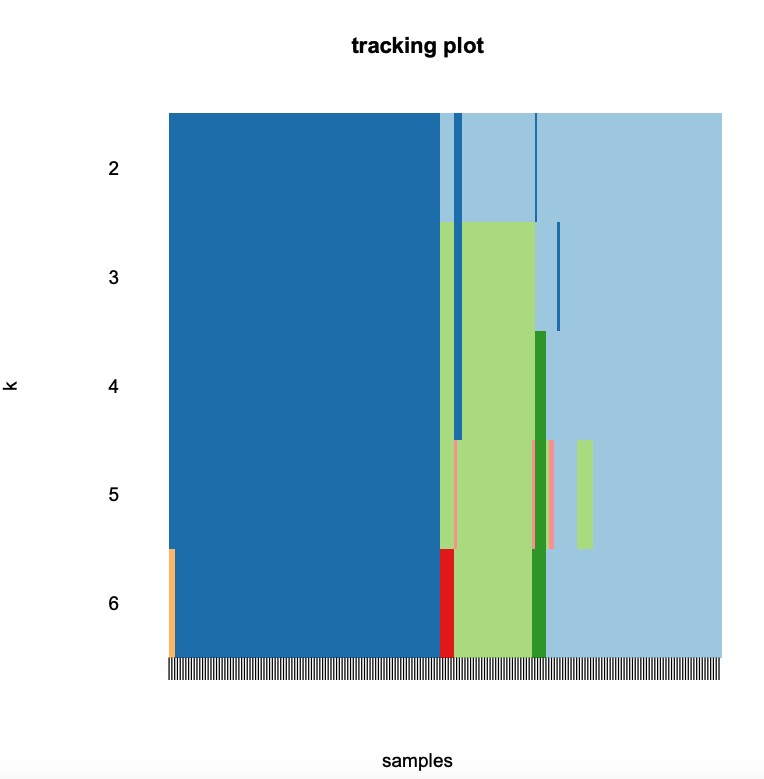
(1)Tracking Plot:此图下方的黑色条纹表示样品,展示的是样品在k取不同的值时,归属的分类情况,不同颜色的色块代表不同的分类。取不同k值前后经常改变颜色分类的样品代表其分类不稳定。若分类不稳定的样本太多,则说明该k值下的分类不稳定。
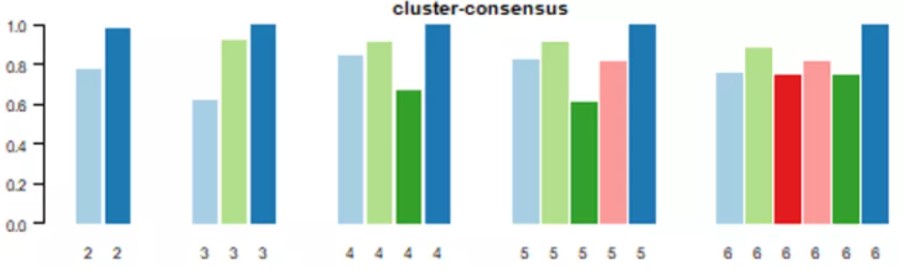
(2)Cluster-Consensus Plot:此图展示的是不同k值下,每个分类的cluster-consensus value(该簇中成员pairwise consensus values的均值)。该值越高(低)代表稳定性越高(低)。可用于判断同一k值下以及不同k值之间cluster-consensus value的高低。
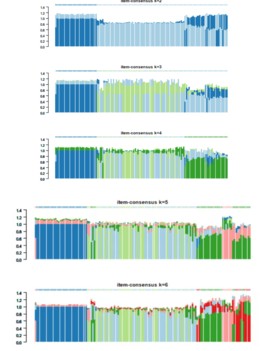
 (3)item-Consensus Plot:纵坐标代表Item-consensus values。k值不同时,每个样本都会有一个对应不同簇的item-consensus values。竖条代表每一个样本,竖条的高度代表该样本的总item-consensus values。每个样本的上方都有一个小叉叉,小叉叉的颜色代表该样本被分到了哪一簇。从这张图,可以看到每个样本的分类是否足够“纯净”,从而帮助决定k值,例如当k=6时,样本的分类变得没有那么纯净,说明k=5才是合适的。
(3)item-Consensus Plot:纵坐标代表Item-consensus values。k值不同时,每个样本都会有一个对应不同簇的item-consensus values。竖条代表每一个样本,竖条的高度代表该样本的总item-consensus values。每个样本的上方都有一个小叉叉,小叉叉的颜色代表该样本被分到了哪一簇。从这张图,可以看到每个样本的分类是否足够“纯净”,从而帮助决定k值,例如当k=6时,样本的分类变得没有那么纯净,说明k=5才是合适的。
- 发表于 2021-05-23 20:44
- 阅读 ( 20675 )
- 分类:TCGA
