geo_gene_exp_download.r GEO数据自动下载与整理
geo_download.r GEO数据下载
使用方法:
$Rscript /share/work/huangls/piplines/omicsclass/tcga_geo/scripts/geo_gene_exp_download.r -h
usage: /share/work/huangls/piplines/omicsclass/tcga_geo/scripts/geo_gene_exp_download.r
[-h] -g gse [-f func] [-p palette] [-G gpl] [-s Gene Symbol] [-x]
[--log2] [-o outdir] [-H height] [-W width]
download GEO data ; https://www.omicsclass.com/article/1492
optional arguments:
-h, --help show this help message and exit
-g gse, --gse gse GEO Series Accession [required]
-f func, --func func dup gene name expression select func: mean max median
[default max]
-p palette, --palette palette
A palette name from RColorbrewer [default Accent]
-G gpl, --gpl gpl GPL file for annotation [default None]
-s Gene Symbol, --symbol Gene Symbol
Gene Symbol column name [default Gene Symbol]
-x, --no.xaxis not show x axis sample name [default False]
--log2 whether do log2 normalize [optional, default: False]
-o outdir, --outdir outdir
output file directory [default /share/nas1/huangls/pro
ject/zx-20210914-383-gwas_cfdr/gene_exp]
-H height, --height height
the height of pic inches [default 8]
-W width, --width width
the width of pic inches [default 10]
使用举例:
Rscript /share/work/huangls/piplines/omicsclass/tcga_geo/scripts/geo_gene_exp_download.r -g GSE7429
Rscript /share/work/huangls/piplines/omicsclass/tcga_geo/scripts/geo_gene_exp_download.r -g GSE43488 -G GPL13667-15572.txt
结果输出:
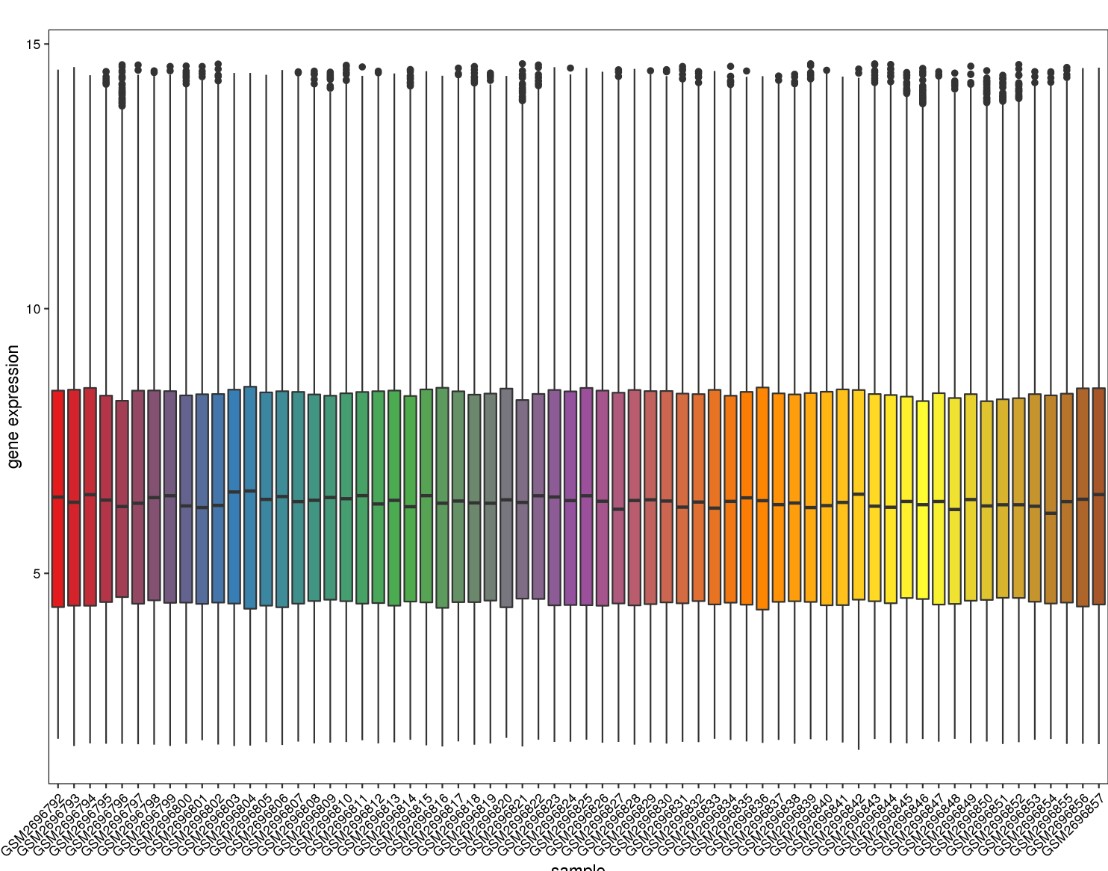
数据整理说明
探针与基因对应:
1.一个基因对应多个探针: 取均值,最大值,等
2.一个探针多个基因,取第一个基因名字
表达量是否需要重新标准化:
可以通过boxplot函数观察一下样本表达丰度值的分布是否整齐进行判断
是否需要log2:根据数据值的大小:
如果表达丰度的数值在50以内,通常是经过log2转化的。如果数字在几百几千,则是未经转化的。
使用过程中常见问题:
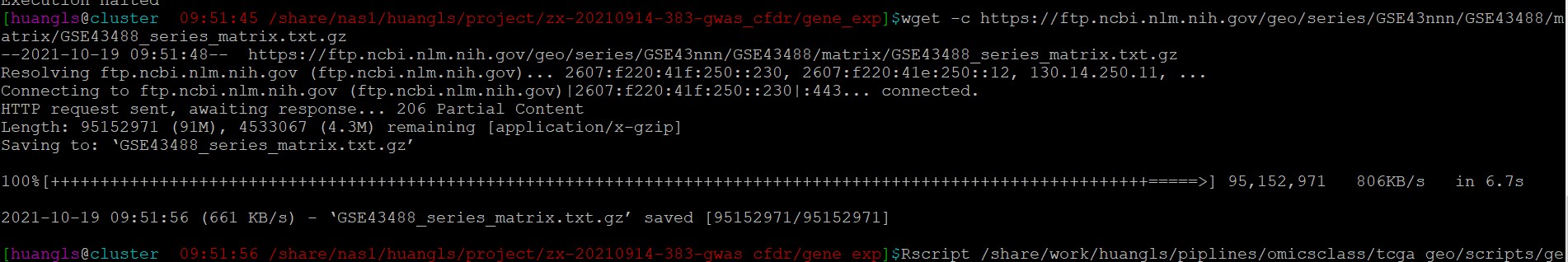
1. 如果中通下载中断 可以用wget -c 下载完成之后再运行该命令:
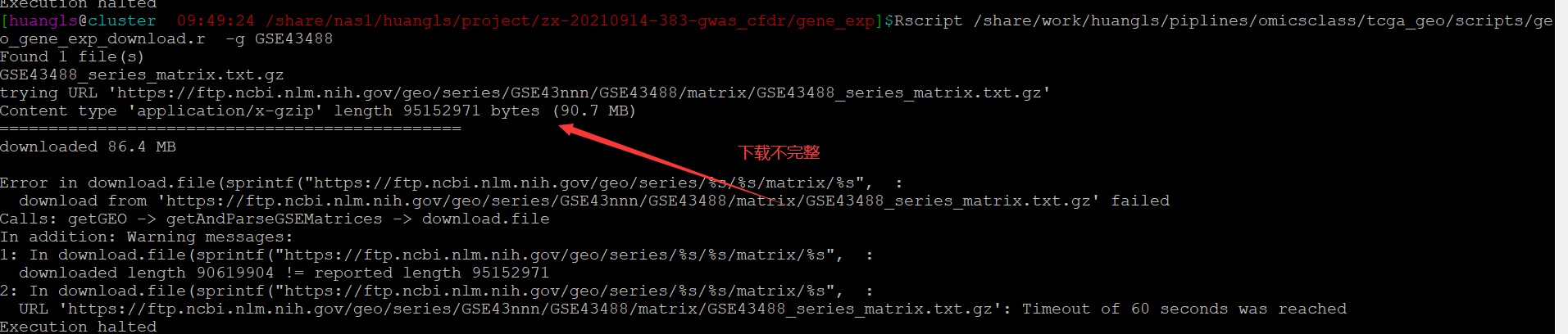 wget -c 继续下载
wget -c 继续下载
可以自己下载GPL文件, GPL 文件应去掉含有#号的行,第一列必须是探针ID,里面必须有 "Gene Symbol" (注意大小写)列不同的基因用 /// 分割:
示例:
Rscript $scripts/geo_gene_exp_download.r -g GSE20680 -o GSE20680 --skip 28
Rscript $scripts/geo_gene_exp_download.r -g GSE20681 -o GSE20681 --skip 28
- 发表于 2021-06-18 17:06
- 阅读 ( 3103 )
- 分类:GEO
