immune_compare_stat.r 免疫细胞表达差异比较
immune_compare_stat.r 免疫细胞表达差异比较
使用说明:
$ Rscript $scriptdir/immune_compare_stat.r -h
usage: /work/STAD_immu_demo1/scripts/immune_compare_stat.r [-h] -i filepath -v
celltype
[celltype ...] -m
filepath -g group
[-b groupby]
[-o path]
[-n prefix]
t.test annova and wilcox.test: https://www.omicsclass.com/article/1496
optional arguments:
-h, --help show this help message and exit
-i filepath, --input filepath
input alpha diversity celltype file[required]
-v celltype [celltype ...], --celltype celltype [celltype ...]
which celltype to compare : "B cell" ...[required]
-m filepath, --metadata filepath
input metadata file path[required]
-g group, --group group
group name from metadata to test[required]
-b groupby, --groupby groupby
main group name from metadata to test [default NULL]
-o path, --outdir path
output file directory [default cwd]
-n prefix, --name prefix
out file name prefix [default demo]
使用示例:
Rscript $scriptdir/immune_compare_stat.r -i immu/ssgsea.res.tsv \
-m metadata.group.tsv -g subtype.hclust \
--celltype "B cells" "T cells" -o immu -n ssgse
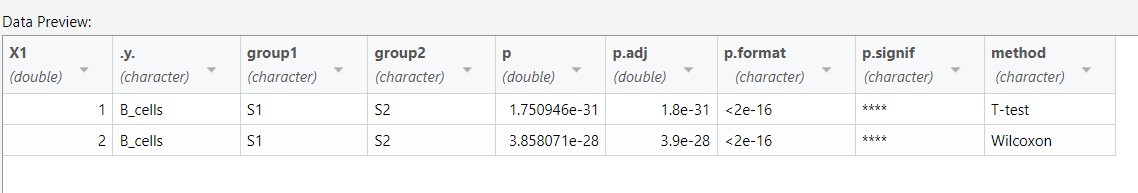
结果:
- 发表于 2021-06-22 12:05
- 阅读 ( 2111 )
- 分类:TCGA
