indel vcf文件转化为SNPT输入文件
如何将indel vcf文件转换为SNPT软件的输入文件。
SNP指纹图谱软件SNPT 这篇文章里介绍了SNPT软件,这里记录一下如何将indel vcf文件转换为SNPT软件的输入文件。
命令如下:
perl /share/work/wangq/script/vcf/vcf_SNPT.indel.pl -vcf clean.vcf -o SNPT.input.txt
vcf_SNPT.indel.pl脚本如下:
#!/share/nas2/genome/bin/perl -w
#use strict;
#use warnings;
use Getopt::Long;
use List::Util qw(shuffle);
use Data::Dumper;
use FindBin qw($Bin $Script);
use File::Basename qw(basename dirname);
use Cwd qw(abs_path);
my $version="1.0.0";
my $BEGIN_TIME=time();
my @Original_ARGV=@ARGV;
# ==============================================================
# Get Options
# ==============================================================
my ($infile, $indexOut, $p);
GetOptions(
"help|?" =>\&USAGE,
"vcf:s"=>\$infile,
"o:s"=>\$out
) or &USAGE;
&USAGE unless ($infile and $out);
sub USAGE {#
my $usage=<<"USAGE";
Program: $0
Version: $version
==================================================================================================
Discription:
This script is used to calculate thresold for BSA using SNP-INDEX method
==================================================================================================
Usage:
Options:
-vcf <str> required vcf list file
-o <str> required output file
USAGE
print $usage;
exit;
}
#===============================================================
# Default optional value
#===============================================================
open (IN, "<$infile") || die "$infile: $!\n";
open (OUT, ">$out") || die "$out: $!\n";
my @sample;
my %snp;
my @indel;
while(<IN>){
chomp;
next if(/^##/);
my ($chr,$pos,$id,$ref,$alt,$qual,$filter,$info,$format,@line) = split(/\t/,$_);
if(/^#/){
@sample = @line;
next;
}
my @array = $_=~/\t([\.0123][\/\|][\.0123])/g;
if(@array != @sample ){
print "SNP $chr,$pos length not eq.\n";
next;
}
my $chr_indel= "$chr-$pos";
push(@indel,$chr_indel);
for(my$i =0; $i<@line ;$i++){
my $str;
if($array[$i] eq "0/0" || $array[$i] eq "0|0" ){
$str = "A";
}elsif($array[$i] eq "0/1" || $array[$i] eq "0|1" ){
$str = "C";
}elsif($array[$i] eq "1/1" || $array[$i] eq "1|1" ){
$str = "G";
}else{
$str = "T";
}
$snp{$chr_indel}{$sample[$i]} = $str;
}
}
close(IN);
print OUT join("\t","Strain",@indel)."\t";
for($i=0; $i < @sample ; $i++){
print OUT "$sample[$i]";
for($j=0; $j < @indel ; $j++){
#print "1";
print OUT "\t$snp{$indel[$j]}{$sample[$i]}";
}
print OUT "\n";
}
close(OUT);
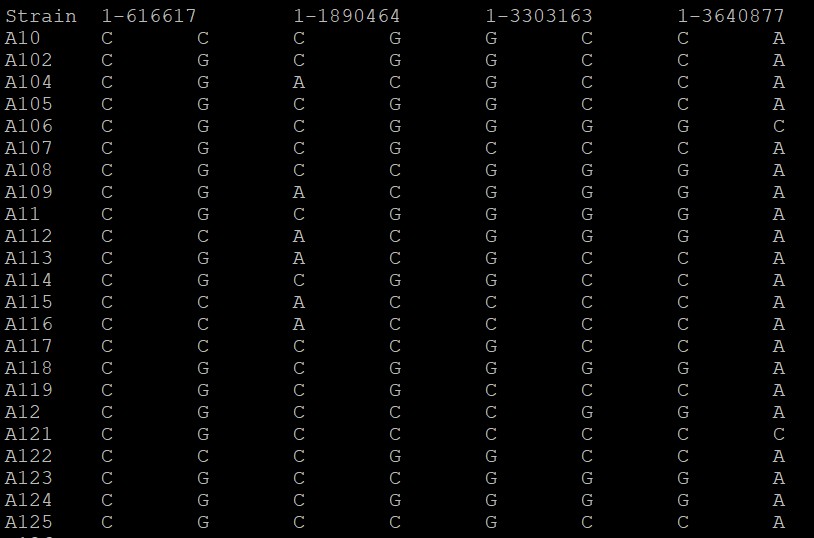
生成文件如下:
此外,我们在网易云课堂上有各种教学视频,有兴趣可以了解一下:
1. 文章越来越难发?是你没发现新思路,基因家族分析发2-4分文章简单快速,学习链接:基因家族分析实操课程
2. 转录组数据理解不深入?图表看不懂?点击链接学习深入解读数据结果文件,学习链接:转录组(有参)结果解读;转录组(无参)结果解读
3. 转录组数据深入挖掘技能-WGCNA,提升你的文章档次,学习链接:WGCNA-加权基因共表达网络分析
4. 转录组数据怎么挖掘?学习链接:转录组标准分析后的数据挖掘
6. 更多学习内容:linux、perl、R语言画图,更多免费课程请点击以下链接:
- 发表于 2021-09-30 10:26
- 阅读 ( 1720 )
- 分类:软件工具
