ssgsea_enrich_diff.r GSVA富集分析
R包GSVA富集分析
使用方法:
Rscript ../scripts/ssgsea_enrich_diff.r -h
usage: ../scripts/ssgsea_enrich_diff.r [-h] -g GMTFILE -i EXPR -m META
[-n GROUP_NAME] [--log2] [-t method]
[-k kcdf] [--group1 GROUP1]
[--group2 GROUP2] [-p PVALUECUTOFF]
[--no_diff] [-o OUTDIR] [-f PREFIX]
Gene set variation analysis (GSVA):https://www.omicsclass.com/article/1586
optional arguments:
-h, --help show this help message and exit
-g GMTFILE, --gmtfile GMTFILE
GSEA gmtfile function class file[required]
-i EXPR, --expr EXPR Input gene expression file path[required]
-m META, --meta META Input the clinical information file path that contains
the grouping[required]
-n GROUP_NAME, --group_name GROUP_NAME
Specifies the column name that contains grouping
information[optional,default:m6acluster]
--log2 Whether to perform log2
processing[optional,default:False]
-t method, --method method
Method to employ in the estimation of gene-set
enrichment scores per sample. By default this is set
to gsva (Hänzelmann et al, 2013) and other options are
ssgsea (Barbie et al, 2009), zscore (Lee et al, 2008)
or plage (Tomfohr et al, 2005). The latter two
standardize first expression profiles into z-scores
over the samples and, in the case of zscore, it
combines them together as their sum divided by the
square-root of the size of the gene set, while in the
case of plage they are used to calculate the singular
value decomposition (SVD) over the genes in the gene
set and use the coefficients of the first right-
singular vector as pathway activity profile[default
gsva]
-k kcdf, --kcdf kcdf Character string denoting the kernel to use during the
non-parametric estimation of the cumulative
distribution function of expression levels across
samples when method="gsva". By default,
kcdf="Gaussian" which is suitable when input
expression values are continuous, such as microarray
fluorescent units in logarithmic scale, RNA-seq log-
CPMs, log-RPKMs or log-TPMs. When input expression
values are integer counts, such as those derived from
RNA-seq experiments, then this argument should be set
to kcdf="Poisson"[default Gaussian]
--group1 GROUP1 Designate the first group[optional,default C1]
--group2 GROUP2 Designate the second group[optional,default C2]
-p PVALUECUTOFF, --pvalueCutoff PVALUECUTOFF
pvalue cutoff on enrichment tests to
report[optional,default:0.05]
--no_diff No screening was performed based on the difference
analysis results[optional,default:False]
-o OUTDIR, --outdir OUTDIR
output file directory[optional,default cwd]
-f PREFIX, --prefix PREFIX
out file name prefix[optional,default kegg]
参数说明:
-g 参考基因集,如从MSigDB下载的KEGG基因集c2.cp.kegg.v6.2.symbols.gmt
| KEGG_GLYCOLYSIS_GLUCONEOGENESIS | http://www.broadinstitute.org/gsea/msigdb/cards/KEGG_GLYCOLYSIS_GLUCONEOGENESIS | ACSS2 | GCK |
| KEGG_CITRATE_CYCLE_TCA_CYCLE | http://www.broadinstitute.org/gsea/msigdb/cards/KEGG_CITRATE_CYCLE_TCA_CYCLE | IDH3B | DLST |
| KEGG_PENTOSE_PHOSPHATE_PATHWAY | http://www.broadinstitute.org/gsea/msigdb/cards/KEGG_PENTOSE_PHOSPHATE_PATHWAY | RPE | RPIA |
-i 基因表达矩阵
| ID | TCGA-A3-3319-01A-02R-1325-07 | TCGA-A3-3323-01A-02R-1325-07 |
| YTHDC2 | 16.5128725081007 | 20.6535652352011 |
| ELAVL1 | 44.3876796198438 | 31.8729000784291 |
-m 包含样本分组信息的样本信息文件
| barcode | patient | sample |
| TCGA-BP-4766-01A-01R-1289-07 | TCGA-BP-4766 | TCGA-BP-4766-01A |
| TCGA-A3-3352-01A-01R-0864-07 | TCGA-A3-3352 | TCGA-A3-3352-01A |
--log2 是否对基因表达矩阵进行log2转换
-n 指定样本信息文件中分组信息的列名
--group1 --group2 指定分组组名
-t 指定用于估计基因集的方法,默认为gsva
-k 指定gsva函数中的kcdf参数,使用read count数据时一般设为“Poisson”,使用log后的TPM等数据时一般就用默认值“Gaussian”
-p 指定p的阈值
--no_diff GSVA会先将表达矩阵转换成富集分数矩阵,然后再通过差异表达分析筛选富集结果,设置这个参数则不对富集结果进行筛选
使用举例:
Rscript ../scripts/ssgsea_enrich_diff.r -g enrich/c2.cp.kegg.v6.2.symbols.gmt \
-i ../02.sample_select/TCGA-KIRC_gene_expression_TPM_immu.tsv -m metadata_group.tsv \
-n m6acluster --log2 --group1 C1 --group2 C2 -p 0.05 -o enrich/C1_vs_C2
结果展示:
富集分数矩阵:kegg_es.tsv
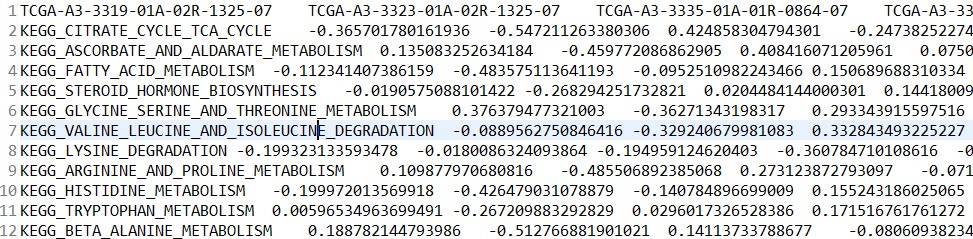
差异分析结果:kegg_diff.tsv
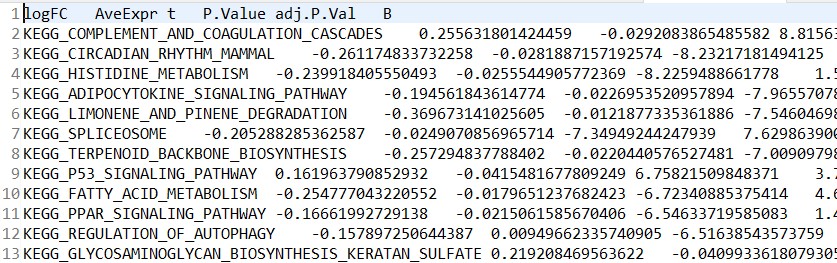
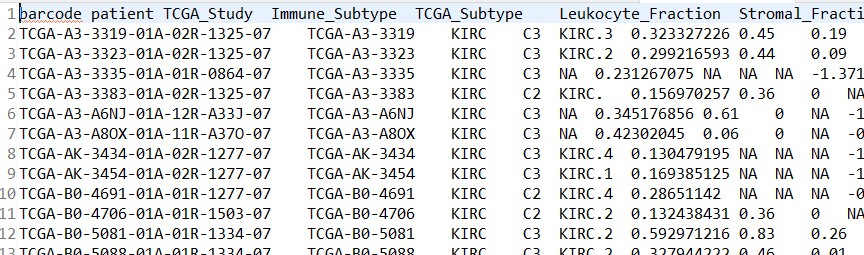
样本信息文件:kegg_meta.tsv
- 发表于 2021-10-29 16:09
- 阅读 ( 3635 )
- 分类:R
