GWAS模型介绍
GWAS关联分析课程推荐:https://bdtcd.xetslk.com/s/RCGWQ
全基因组关联分析(Genome wide association study,GWAS)是对多个个体在全基因组范围的遗传变异(标记)多态性进行检测,获得基因型,进而将基因型与可观测的性状,即表型,进行群体水平的统计学分析,根据统计量或显著性 p 值筛选出最有可能影响该性状的遗传变异(标记),挖掘与性状变异相关的基因。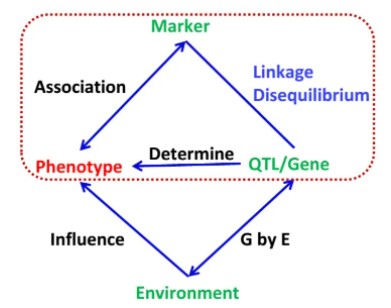
GWAS是传统双亲种群绘图的替代方法,目前广泛用于植物、动物、模式物种和人类,与传统的QTL定位相比,GWAS优点包括更高的分辨率、研究材料来源广泛,可捕获的变异丰富,无需构建遗传群体节省时间。
GWAS是传统双亲种群绘图的替代方法,目前广泛用于植物、动物、模式物种和人类,与传统的QTL定位相比,GWAS优点包括更高的分辨率、研究材料来源广泛,可捕获的变异丰富,无需构建遗传群体节省时间。
GWAS分析模型介绍
GWAS 分析一般会构建回归模型检验标记与表型之间是否存在关联。GWAS中的零假设(H0 null hypothesis)是标记的回归系数为零, 标记对表型没有影响。备择假设(H1,也叫对立假设,Alternative Hypothesis)是标记的回归系数不为零,SNP和表型相关。GWAS中的模型主要分为两种:
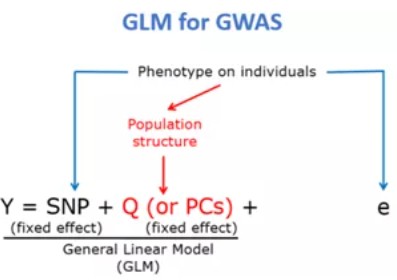
一般线性模型GLM(General Linear Model):y = Xα + Zβ + e
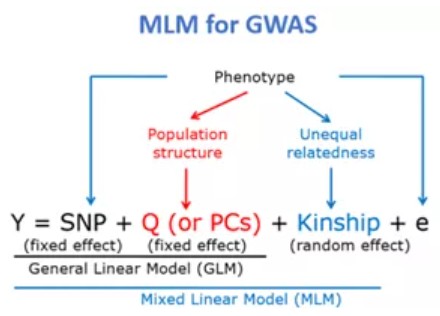
混合线性模型MLM(Mixed Linear Model):y = Xα+ Zβ + Wμ+ e
y: 所要研究的表型性状;
Xα:固定效应(Fixed Effect),影响y的其他因素,主要指群体结构;
Zβ:标记效应(Marker Effect SNP);
Wμ:随机效应(RandomEffect),这里一般指个体的亲缘关系。
e: 残差
GWAS分析一直需要解决两个问题,一个是随着测序数据量的不断增加,计算速度已经是影响GWAS分析的一个重要问题。二是统计的准确率能不能再增加一些。因此发展出了很多其他的模型,请看下面的图形,图中的河流代表GWAS分析方法的不断发展,从由上角的Q模型到最下面的Blink,GWAS分析方法经历了几代人的努力。

GWAS模型详细介绍:
一般线性模型GLM:直接将基因型x和表型y做回归拟合。也可以加入群体结果控制假阳性。
 混合线性模型MLM:GLM模型中,如果两个表型差异很大,但群体本身还含有其他的遗传差异(如地域等),则那些与该表型无关的遗传差异也会影响到相关性。MLM模型可以把群体结构的影响设为协方差,把这种位点校正掉。此外,材料间的公共祖先关系也会导致非连锁相关,可加入亲缘关系矩阵作为随机效应来矫正。
混合线性模型MLM:GLM模型中,如果两个表型差异很大,但群体本身还含有其他的遗传差异(如地域等),则那些与该表型无关的遗传差异也会影响到相关性。MLM模型可以把群体结构的影响设为协方差,把这种位点校正掉。此外,材料间的公共祖先关系也会导致非连锁相关,可加入亲缘关系矩阵作为随机效应来矫正。
随着二代测序技术的发展,基因分型变得越来越容易,用于关联分析的样本量和标记数不断增大,原始的MLM模型求解所耗的时间可以用mpn3来表示(m为标记数目,p为求解过程的迭代次数,n为样本数),可见,随着样本量的增加,每迭代一步,计算时间都会以样本3次方增长,这使得计算的时间变得非常长。为解决这一问题,Zhang等提出了P3D(population parameters previously determined)和压缩混合线性模型(compressed MLM, CMLM),并将这两种方法整合到TASSEL软件中,大大提高了计算效率,检测功效也得到提高。P3D减少了重复计算方差组分的次数;CMLM通过聚类减少了实际参与计算的样本数。考虑到8种聚类方法和3种组间亲缘关系算法的组合可能得到不同的结果,检测最优组合的优化压缩混合线性模型(enriched CMLM, ECMLM)被提出,并整合在GAPIT软件中。
CMLM压缩混合线性模型:MLM的矫正过于严格,会把一些真实相关的SNP标记也过滤掉,因此CMLM模型目的是重新检测到那些假阴性SNP标记。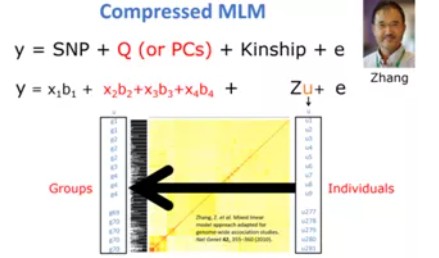
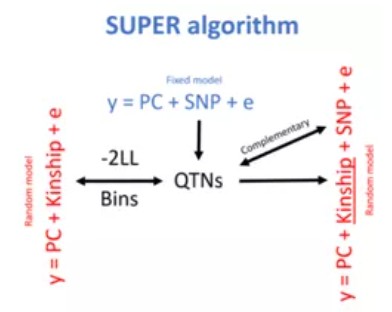
SUPER:CMLM应该选择哪些SNP来计算亲缘关系矩阵,答案是使用所有跟表型相关的SNP(且排除了检测到的那个SNP)来构建亲缘关系矩阵的效果最好,这就是SUPER(Settlement of Kinship Under Progressively Exclusive Relationship, 逐步排他性亲缘关系解决方案)。
 FarmCPU:GWAS的瓶颈一是计算速度,二是统计准确性。FarmCPU能提升速度和准确性,首先把随机效应的亲缘关系矩阵(Kinship)转换为固定效应的关联SNP矩阵(S矩阵/QTNs矩阵),使计算速度大大加快;再利用QTN矩阵当做协变量,重新做关联分析,提升准确率。
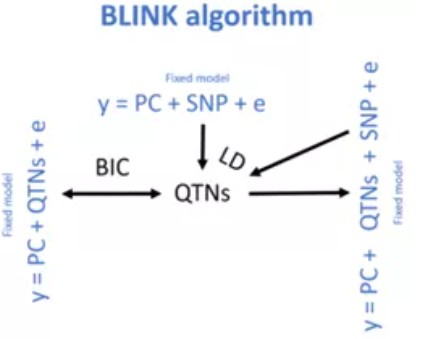
FarmCPU:GWAS的瓶颈一是计算速度,二是统计准确性。FarmCPU能提升速度和准确性,首先把随机效应的亲缘关系矩阵(Kinship)转换为固定效应的关联SNP矩阵(S矩阵/QTNs矩阵),使计算速度大大加快;再利用QTN矩阵当做协变量,重新做关联分析,提升准确率。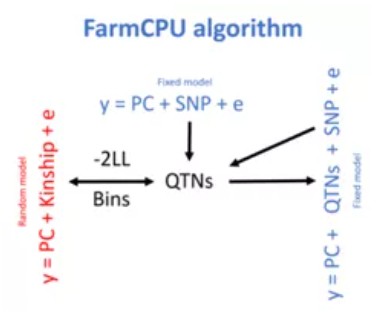 Blink:Blink是进阶版FarmCPU,也是为提高速度和准确率。先用上方的GLM模型获得QTNs,然后用右侧的GLM以QTNs当做协变量进行SNP检测,得到的SNP根据LD信息确定QTNs的信息(根据染色体实际位置来选择对应的bin大小),进而利用左侧的GLM以BIC(Bayesianinformation criterion)策略进行QTNs准确性检测,排除假设错误的部分,保留真实的QTNs,不断循环这一过程,直到检测到所有关联SNP(即QTNs)。
Blink:Blink是进阶版FarmCPU,也是为提高速度和准确率。先用上方的GLM模型获得QTNs,然后用右侧的GLM以QTNs当做协变量进行SNP检测,得到的SNP根据LD信息确定QTNs的信息(根据染色体实际位置来选择对应的bin大小),进而利用左侧的GLM以BIC(Bayesianinformation criterion)策略进行QTNs准确性检测,排除假设错误的部分,保留真实的QTNs,不断循环这一过程,直到检测到所有关联SNP(即QTNs)。
其他模型:
Kang等通过减少需要估计的方差组分的个数和简化矩阵逆运算的过程,提出了EMMA模型,在此基础上,通过避免重复估计多基因方差和误差方差,提出了EMMAX算法,并开发了EMMAX软件,进一步提高了计算速度,但由于多基因方差和误差方差的比值固定,EMMA和EMMAX都属于近似算法,而Zhou等提出的GEMMA算法为EMMA的精确算法。
参考文献:
Price, A. L., Patterson, N. J., Plenge, R. M., Weinblatt, M. E., Shadick, N. A., et al. (2006). Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 38 (8), 904–909. doi: 10.1038/ng1847
Yu, J., Pressoir, G., Briggs, W. H., Vroh, B. I., Yamasaki, M., Doebley, J. F., et al. (2006). A unifed mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat. Genet. 38, 203–208. doi: 10.1038/ng1702
Zhang, Z., Ersoz, E., Lai, C. Q., Todhunter, R. J., Tiwari, H. K., Gore, M. A., et al. (2010). Mixed linear model approach adapted for genome-wide association studies. Nat. Genet. 42, 355–360. doi: 10.1038/ng.546
Li, M., Liu, X., Bradbury, P., Yu, J., Zhang, Y.-M., Todhunter, R. J., et al. (2014). Enrichment of statistical power for genome-wide association studies. BMC Biol. 12, 73. doi: 10.1186/s12915-014-0073-5
Segura, V., Vilhjálmsson, B. J., Platt, A., Korte, A., Seren, Ü., Long, Q., et al. (2012). An efficient multi-locus mixed-model approach for genome-wide association studies in structured populations. Nat. Genet. 44, 825–830. doi: 10.1038/ng.2314
Wang, Q., Tian, F., Pan, Y., Buckler, E. S., Zhang, Z. (2014). A SUPER powerful method for genome wide association study. PLoS ONE 9, e107684. doi: 10.1371/journal.pone.0107684
Liu, X., Huang, M., Fan, B., Buckler, E. S., Zhang, Z. (2016). Iterative usage of fixed and random effect models for powerful and efficient genome-wide association studies. PLoS Genet. 12 (2), e1005767. doi: 10.1371/journal.pgen.1005767
延伸阅读
- 颠覆认知,百万样品GWAS的杰作!
- 群体进化和GWAS文章没那么难发!
- Nature Biotechnology:基于GWAS与群体进化分析挖掘大豆驯化及改良相关基因
- 必备技能:引物设计(SSR、SNP、InDel)
- 必备技能:富集分析-GO/KEGG气泡图和柱状图的绘制
- 必备技能:SRA数据下载、处理、blast比对
- 发表于 2021-11-15 13:17
- 阅读 ( 15370 )
- 分类:GWAS
