IF:8.8|TCGA+ceRNA+肝癌文献解析
ceRNA,中文名竞争性內源RNA。和ncRNA等概念不同,ceRNA并不是代表某种特定类型的RNA,而是一种调控机制,指生物体内复杂的转录调控网络中的RNA,主要包括蛋白编码基因mRNA、长链非编码RNA(lncRNA)、环状RNA(circRNA)、假基因(pseudogenes)等。
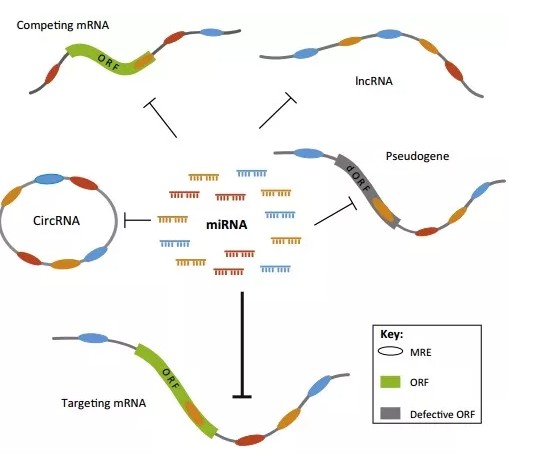 如上图:miRNA可通过抑制靶基因mRNA的翻译,或促进mRNA降解来起到负调控的作用;circRNA,能够像海绵一样吸附、结合miRNA,使其不能发挥负调控的作用,从而促进靶基因的表达;lncRNA可以与miRNA互作,作为ceRNA并吸附miRNA,抑制miRNA的作用,促进mRNA表达。在这个过程中miRNA和circRNA起到拮抗的作用:miRNA水平升高抑制靶基因表达,circRNA水平升高则促进靶基因表达。其中的几个主角:lncRNA 、 circRNA、miRNA、靶基因mRNA共同构成了ceRNA调控网络。一个靶基因可能被多个miRNA、circRNA/lncRNA同时调控,所以ceRNA调控网络可能非常的庞大。
如上图:miRNA可通过抑制靶基因mRNA的翻译,或促进mRNA降解来起到负调控的作用;circRNA,能够像海绵一样吸附、结合miRNA,使其不能发挥负调控的作用,从而促进靶基因的表达;lncRNA可以与miRNA互作,作为ceRNA并吸附miRNA,抑制miRNA的作用,促进mRNA表达。在这个过程中miRNA和circRNA起到拮抗的作用:miRNA水平升高抑制靶基因表达,circRNA水平升高则促进靶基因表达。其中的几个主角:lncRNA 、 circRNA、miRNA、靶基因mRNA共同构成了ceRNA调控网络。一个靶基因可能被多个miRNA、circRNA/lncRNA同时调控,所以ceRNA调控网络可能非常的庞大。
解读文献

研究背景
肝细胞癌(HCC)是危害人类健康的最致命的恶性肿瘤之一。越来越多的证据强调竞争性内源RNA(ceRNA)调控网络在各种人类癌症中的关键作用。然而,HCC中ceRNA网络的复杂性和行为特征仍不清楚。在这项研究中,旨在阐明磷酸酶和张力蛋白同源物(PTEN)相关的ceRNA调节网络,并确定与HCC相关的潜在预后标志物
ceRNA网络构建分析流程
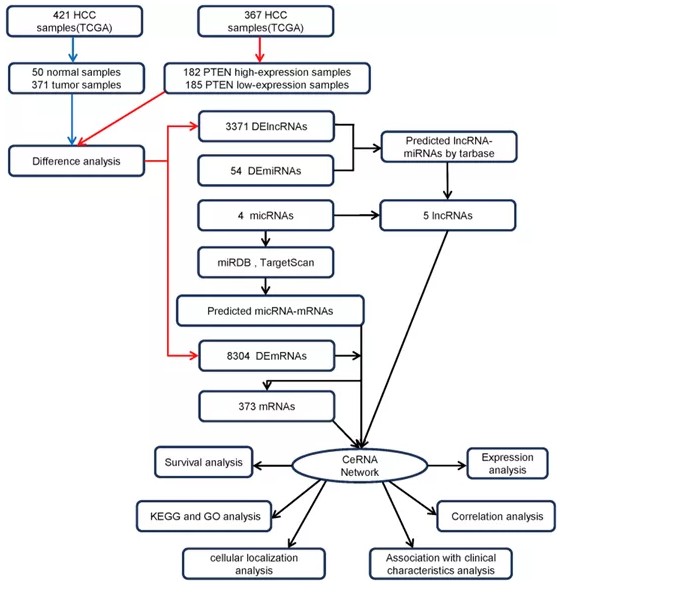
数据:TCGA数据库人类HCC表达谱数据以及临床信息(LIHC),表达数据包括lncRNA、mRNA以及miRNA;GEO基因表达谱-GSE410804作为验证数据集靶基因预测数据库:tarbase、targetscan、miRDB数据分析思路:1.样本按照两组分组类型(PTEN高表达/低表达、肿瘤组织/正常组织)分别进行差异表达分析,获得各自的差异mRNA、lncRNA以及miRNA。2.对两组差异lncRNA取交集,获得的lncRNA在tarbase数据库中预测下游miRNA;预测到的miRNA与差异miRNA取交集,获得的miRNA在miRDB、TargetScan数据库中预测靶mRNA;预测到的靶mRNA与差异mRNA取交集。3.最终获得的差异lncRNA、miRNA、mRNA以及他们之间的调控关系,通过Cytoscape构建ceRNA网络,并通过其中的Cytohubba插件进行分析筛选hub ceRNA网络。4.对所筛选得到的子网络进行相关性分析,km分析,细胞定位分析,根据分析结果进一步选择潜在的预后模型。
5.模型中的RNA结合临床特征进行单因素、多因素cox回归分析,从而获得具有独立预后效果的RNA。
6.探究具有独立预后效果的RNA与免疫浸润之间的关系,并通过富集分析来确定其在HCC中的潜在功能。
探究PTEN在HCC中的抑癌作用和预后价值
首先探讨PTEN在肝细胞癌中的作用,(A)通过人类蛋白质图谱数据库(HPA)发现PTEN在肝癌中低表达,在正常组织中过表达。(B)HPA的免疫组化染色结果也证实了相似的PTEN异常表达。(C)由于PTEN在肿瘤组织的异常低表达,研究了PTEN在肝细胞癌患者中表达的临床意义,绘制了km曲线,结果表明PTEN的异常低表达与患者生存率低显著相关。随后,为了解PTEN在肿瘤样本中异常低表达的潜在机制,(D)通过cBioPortal分析PTEN的基因组和拷贝数,结果显示TCGA HCC数据集中PTEN基因的缺失,(E)此外PTEN缺失的肝细胞癌组织的mRNA表达低于二倍体PTEN的肝细胞癌组织,(F)同时,PTEN拷贝数值与mRNA表达呈正相关。

鉴定差异mRNA、lncRNA、miRNA
根据以上分析结果,与PTEN相关的ceRNA网络可以作为预测肝细胞癌患者预后的潜在模型。
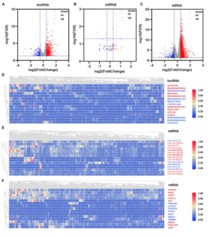
首先采用TCGA数据库鉴定具有PTEN高表达和PTEN低表达的HCC样本之间、以及HCC组织与癌旁组织之间的差异mRNA、lncRNA、miRNA。以p<0.05和|logFC|>0.5作为筛选差异lncRNA的阈值,以p<0.05和|logFC|>0.3作为筛选差异miRNA的阈值,以p<0.005和|logFC|>0.7/0.5作为筛选差异mRNA的阈值。筛选结果:PTEN高低表达之间差异mRNA-1871个(195上调、1676下调)、差异lncRNA-860个(137上调、723下调)、差异miRNA-54个(21上调、33下调);HCC组织与癌旁组织之间差异mRNA-8294个(7059上调、1235下调)、差异lncRNA-3371个(3041上调、330下调)、差异miRNA-420个(102上调-318下调)
(A-C)火山图展示PTEN高低表达组之间差异mRNA、差异lncRNA、差异miRNA的分布。(D-F)热图展示15个显著的差异mRNA、差异lncRNA、差异miRNA
构建ceRNA网络
首先对PTEN高/低组之间的差异lncRNA以及癌症组织/正常组织之间的差异lncRNA取交集,根据这些交集lncRNA在TarBase数据库中,识别潜在的靶miRNA;预测到的miRNA与PTEN高/低组之间的差异miRNA以及癌症组织/正常组织之间的差异miRNA的交集miRNA取交集,筛选到4个miRNA;之后使用miRDB以及TargetScan数据库来预测这4个miRNA的下游mRNA,除此之外,为提高预测准确性,仅取两个数据库共同的预测结果。(A)结果显示,在差异mRNA中鉴定到了373个mRNA。最后共有5个lncRNA(4上调、1下调)、4个miRNA(1上调、3下调)、372mRNA用来构建ceRNA网络。之后采用Cytoscape来识别筛选ceRNA网络中的hub网络。(B)结果显示3个lncRNA(DLEU2L、FAM99A、ARRDC1-AS1)、4个miRNA(miR-99a-5p、miR-100-5p、miR-9-5p、miR-125b-5p)、以及6个mRNA(TAOK1、HS3ST3B1、RHOQ、BAZ2A、AGO2、NR6A1)被筛选出来。为了探索hub网络潜在的功能,利用Metascape进行功能富集分析(KEGG、GO),(C)结果显示网络中的mRNA被特异性富集在”蛋白质激酶活性“、”GTP结合酶“、”参与细胞分化的形态发生“

构建并验证核心ceRNA网络、筛选HCC预后模型
为了在HCC中建立具有良好预后价值的核心ceRNA网络,首先分析hub网络中RNA在PTEN高/低组以及癌症组织/正常组织中的表达水平。正如之前所提到的,PTEN高/低组中的RNA表达水平与癌症组织/正常组织相反。结果显示,在PTEN高/低组中,hub网络中RNA表达水平为:1个下调lncRNA(DLEU2L)和2个无差异lncRNA(ARRDC1-AS1、FAM99A)、一个上调miRNA(miR-9-5p)和三个下调miRNA(miR-99a-5p、miR-100-5p、miR-125b-5p)、三个下调mRNA(TAOK1、HS3ST3B1、AGO2)和三个无差异mRNA(RHOQ、BAZ2A、NR6A1);而在癌症组织/癌旁组织中,两个lncRNA(DLEU2L、ARRDC1-AS1)上调和一个lncRNA下调(FAM99A)、一个上调miRNA(miR-9-5p)和三个下调miRNA(miR-99a-5p、miR-100-5p、miR-125b-5p)、五个上调mRNA(TAOK1、RHOQ、BAZ2A、NR6A1、AGO2)和一个下调mRNA(HS3ST3B1)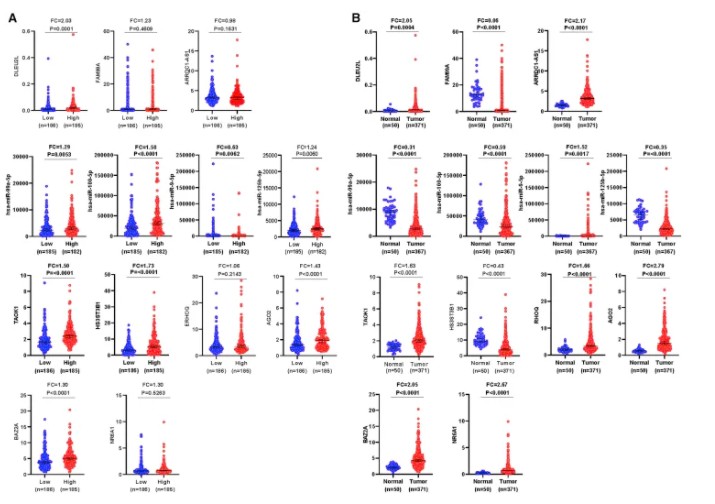 为了确定这些RNA与生存是否相关,使用KM分析对患者进行OS分析。发现一种lncRNA(DLEU2L)、两种miRNA(miR-99a-5p、miR-100-5p)和三种mRNA(TAOK1、HS3ST3B1、RHOQ)与预后相关。
为了确定这些RNA与生存是否相关,使用KM分析对患者进行OS分析。发现一种lncRNA(DLEU2L)、两种miRNA(miR-99a-5p、miR-100-5p)和三种mRNA(TAOK1、HS3ST3B1、RHOQ)与预后相关。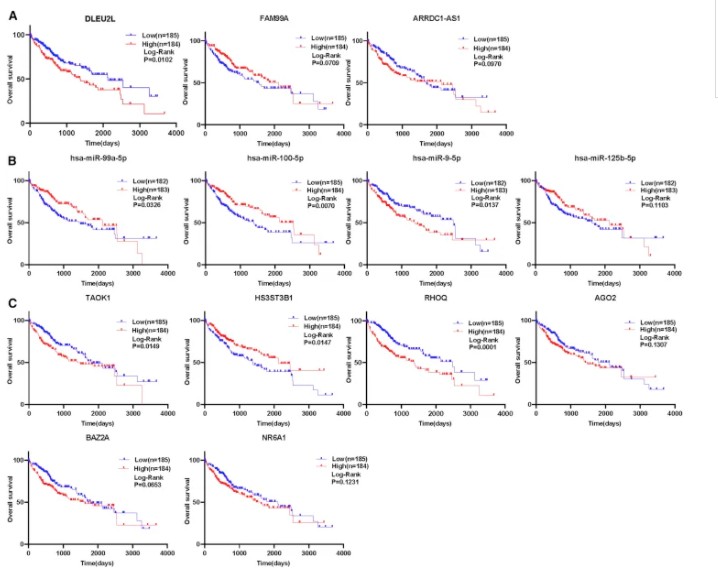 除此之外,由于lncRNA的细胞定位决定潜在机制,通过lncLocator分析三种lncRNA的亚细胞定位。(A)DLEU2L主要分布在细胞质中,其他两个lncRNA分布在细胞质基质中。
除此之外,由于lncRNA的细胞定位决定潜在机制,通过lncLocator分析三种lncRNA的亚细胞定位。(A)DLEU2L主要分布在细胞质中,其他两个lncRNA分布在细胞质基质中。通过以上分析,可推测DLEU2L可能吸附miR-99a-5p/miR-100-5p来提高TAOK1的表达。(B)因此构建了DLEU2L-miR-99a-5p/miR-100-5p-TAOK1 ceRNA网络。(C)Tarbase和TargetScan预测DLEU2L和TAOK1的3端UTR中的靶位点分别与miR-99a-5p/miR-100-5p配对。(D)此外,表达相关性分析表明DLEU2L与TAOK1之间存在正相关性关系。
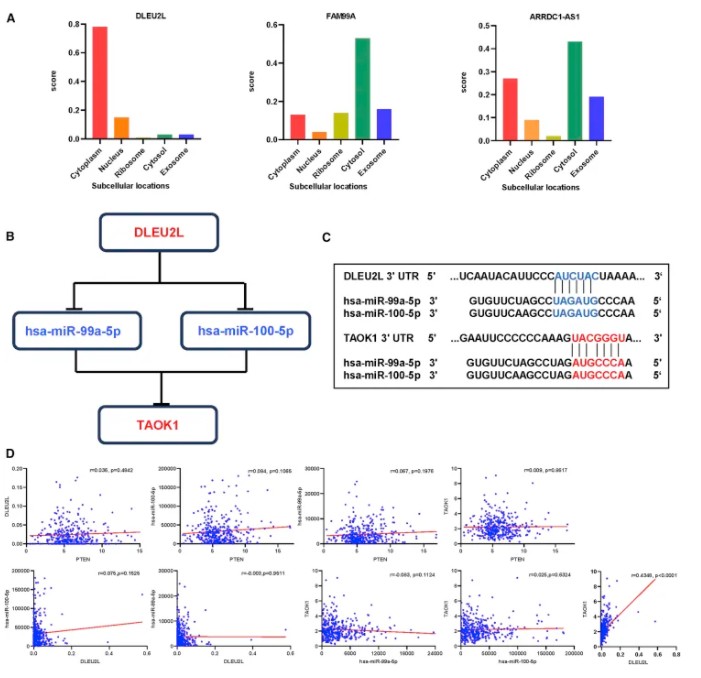 因此选择ceRNA网络中的DLEU2L/TAOK1轴作为潜在预后模型进行分析
因此选择ceRNA网络中的DLEU2L/TAOK1轴作为潜在预后模型进行分析DLEU2L/TAOK1轴在HCC患者中的临床相关性
为了确定DLEU2L/TAOK1的表达水平是否和临床因素相关,对此进行相关性分析。通过单因素和多因素cox回归分析来确定生存相关特征。结果显示在单因素cox回归分析结果中DLEUL2与TAOK1过表达水平与较差预后显著相关,而在多因素cox回归分析中,DLEU2L表达与不良预后无关。因此,TAOK1可能为HCC患者的独立预后因素。TAOK1异常高表达验证
根据GEO数据库中的HCC患者样本中的TAOK1表达进行差异分析,结果显示TAOK1在HCC组织中的表达明显高于正常组织,这与TCGA数据得到的结果一致。TAOK1表达与免疫浸润的相关性
为了评估肿瘤免疫浸润水平与TAOK1表达之间的潜在关系,(A)使用TIMER分析不同免疫细胞中TAOK1的基因拷贝数。(B)分析TAOK1与免疫细胞浸润水平之间的相关性。(C)评估免疫浸润对患者预后的影响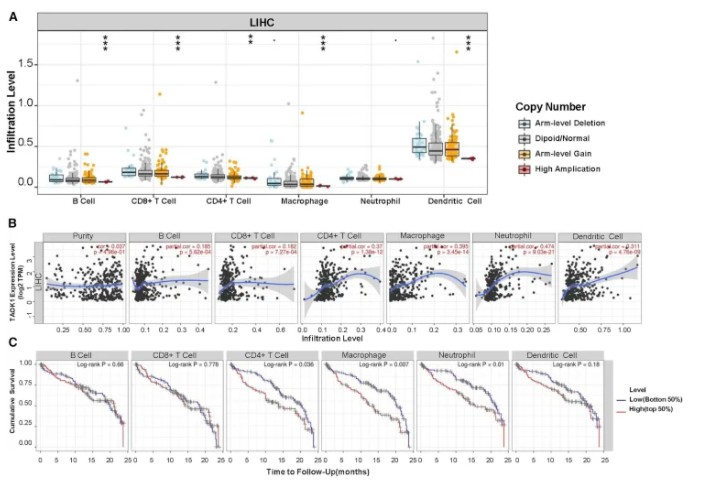

- 发表于 2022-01-07 10:15
- 阅读 ( 3096 )
- 分类:文献解读
