IF=6.68| HCC肝癌单细胞数据挖掘实践
单细胞转录组测序技术的普及,为生命科学的研究提供了全新的手段。基于高通量单细胞转录组测序,既能发现特定组织中可能的细胞类型,也能了解不同细胞组成上的差别,更能通过基因表达图谱剖析不同细胞的基因特征,为理解细胞的功能提供数据支持。
针对单细胞数据的挖掘也使我们从一个全新的角度来解析我们的数据。近年来,关于单细胞数据挖掘的文章相对新颖也容易发高分,这里小编介绍一篇单细胞数据挖掘的SCI文章(影响因子6.68)。
同时,我们生信博士提供高效快捷的筛选marker基因的脚本供大家学习使用。
单细胞数据介绍

首先小编简单介绍下这篇被二次挖掘多次的单细胞数据分析的原文,这篇文章的主要内容如下:
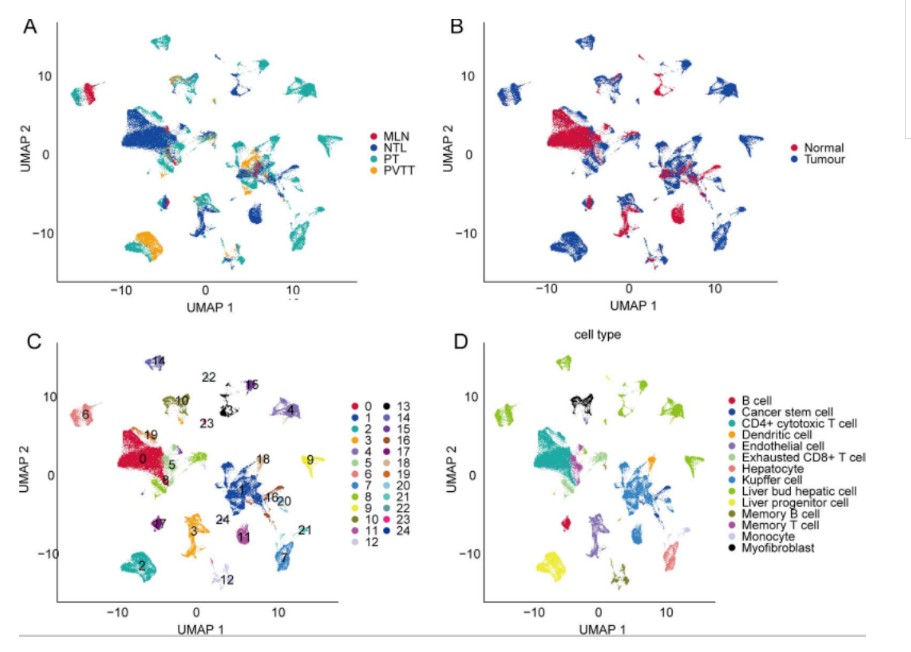
作者选取了10个肝癌病人共21个样本,取样类型包括:primary tumor (T)、portal vein tumor thrombus (P)、metastatic lymph node (L)、non-tumor liver (N);通过单细胞测序共得到28,687 non-tumor liver cells和4,414 primary tumor cells。通过Umap聚类分析得到28个clusters。
通过比较肿瘤样本和非肿瘤样本的聚类结果发现,有些亚类细胞只在肝癌细胞中出现(clusters 8, 13, 16, and 17),其Marker基因为:HCC markers (GPC3, CD24, and MDK)。进一步通过拟时序分析这4个亚类细胞(clusters 8, 13, 16, and 17),得到4个cluster,其中cluster 0是最初始细胞亚类,发现PGLS这个基因在cluster 0中高表达。通过TCGA中的数据,发现这个基因高表达与病人的预后差相关。在后续的湿实验中,作者重点验证了这个基因(通过激活戊糖磷酸途径促进肝细胞癌变),包括敲低、过表达、癌细胞侵袭实验等。
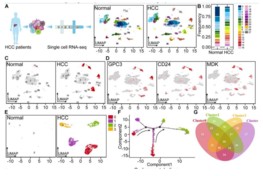
针对以上文章中的数据做二次挖掘所发文章介绍

原文刚刚发布,其数据很快又被利用挖掘发新的文章。原文中关于单细胞的分析内容相对较少,只是分析了其中的三团细胞找到marker基因,再加了后续湿实验验证。单细胞数据没有充分挖掘利用实属可惜。但是,这给我们生信数据挖掘的人一个大好的机会,可以从不同的角度对数据重新进行分析再挖掘。下面介绍这篇文章就是对上篇文章数据进行二次挖掘分析的,其主要的分析思路如下:
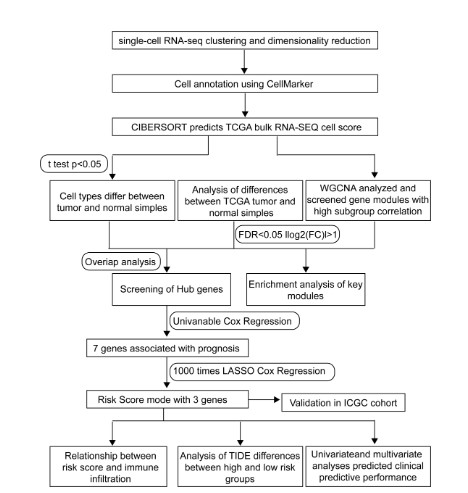
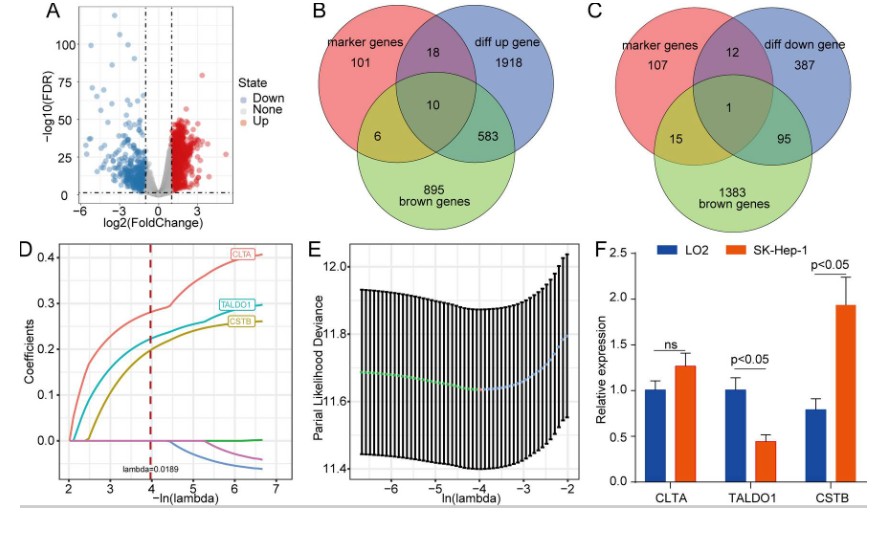
- 单细胞分析结果:单细胞数据标准分析获得25个cluster,利用CellMarker数据库对不同细胞cluster注释;
利用单细胞数据分析得到细胞类型组成,再借助CIBERSORT工具分析TCGA中肝癌数据转录组细胞亚型的组成。结果表明,很多类型的细胞组成在正常组织和肿瘤之间存在差异。作者再结合WGCNA构建共表达网络,发现cluster21 monocyte 与棕色模块相关;
- TCGA肿瘤组织和正常组织的差异基因与棕色模块基因取交集,利用lasso筛选到三个基因 CLTA、TALDO1和CSTB,构建预后模型。
复现单细胞数据分析部分步骤
1. 数据下载数据下载链接:https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE149614单细胞数据一般为高通量测序的数据,原始数据为fastq格式,数据量往往很大。如果公开的数据中有单细胞分析的基因表达矩阵,我们就可以直接用该表达矩阵文件进行后续数据挖掘,不用对原始数据重头分析。正好这个数据也提供了表达数据以及样本信息,这里我们下载counts数据即可。
数据介绍:portal vein tumor thrombus (PVTT),T primary tumor (PT),L metastatic lymph node (MLN),N adjacent non-tumor liver (NTL);分别对应样本数据名称中缩写名称(P、T、L、N)。
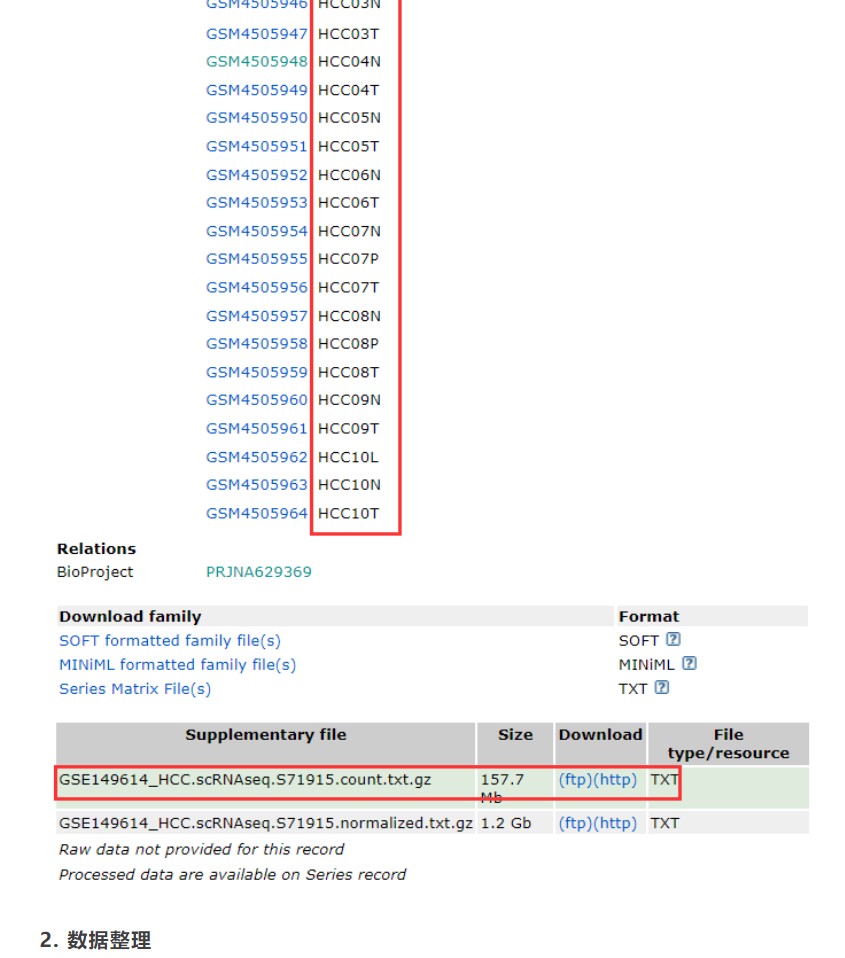
2. 数据整理
单细胞的表达矩阵数据格式如下图所示,我们需要整理不同细胞的metadata数据:每个细胞属于哪个病人,以及属于什么类型的组织等等。有了metadata数据我们才能将细胞分类然后再进行后续的分析。
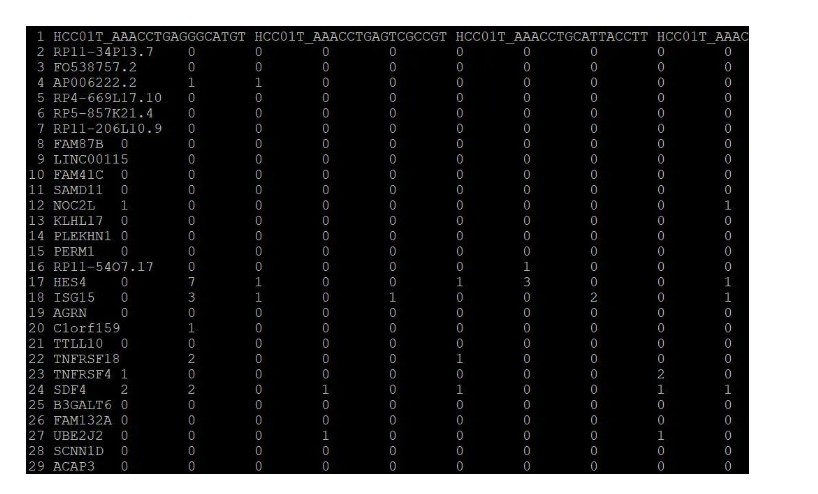
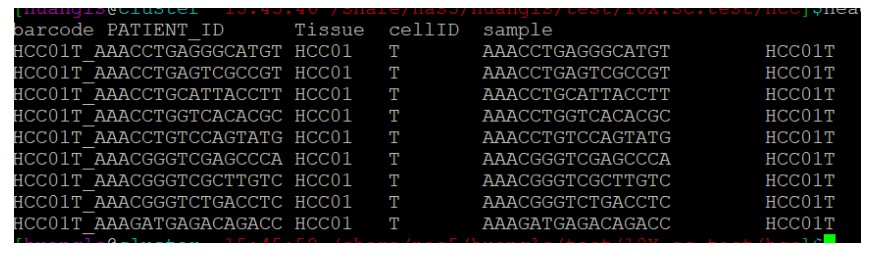
通过观察单细胞的表达矩阵数据(即上图第一行信息),我们可以把第一行信息提取出来,并整理成以下metadata格式数据。整理过程可以用以下shell代码快速实现。
zcat GSE149614_HCC.scRNAseq.S71915.count.txt.gz|head -1|sed 's#\t#\n#g'|perl -e 'while(<>){chomp;/^(HCC\d+)(\w)_([ATCGN]+)$/;print "$_\t$1\t$2\t$3\t$1$2\n"}' |sed '1ibarcode\tPATIENT_ID\tTissue\tcellID\tsample' >metadata.tsv
3. 利用脚本快速的进行单细胞数据分析
3.1 数据读入与质控
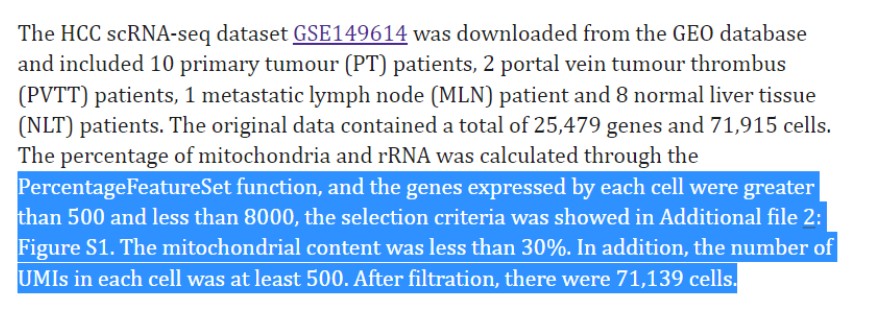
细胞过滤参数:
表达基因数量过滤,500-8000;UMI数量大于500,线粒体基因比例小于30%。具体可以按照以下代码运行:
Rscript scripts/seurat_sc_qc.r -i GSE149614_HCC.scRNAseq.S71915.count.txt.gz \ --metadata metadata.tsv \ --project HCC -p hcc --sep "\t" \ -o 01.read.hcc \ --nGene.min 500 --nGene.max 8000 --percent_mito 30 --nUMI.min 500
数据过滤后结果如下图所示:
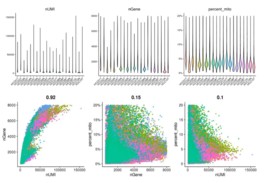
3.2 单细胞聚类分析

单细胞聚类分析的关键参数如上图所示:聚类分析用到的PCA主成分前50轴,高变基因2000个,resolution 分辨率0.1。其中分辨率越高聚类数量越多,为避免聚类结果太多,作者选用0.1的分辨率。以下为运行代码:
###2 单细胞聚类分析 Rscript scripts/seurat_sc.r --resolution 0.1 --pt.size 0.2 \ --rds 01.read.hcc/hcc.afterQC.rds -o 02.cluster -p hcc --high.variable.genes 2000 --dim 50
#2.1查找marker 基因 Rscript scripts/seurat_FindAllMarkers.r -i 02.cluster/hcc.rds \ -o 02.cluster/ -p hcc.allMarker
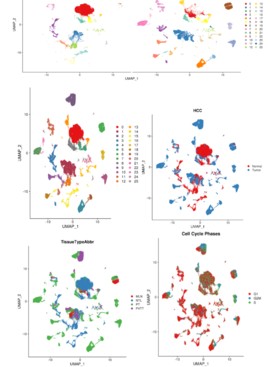
#2.2不同组织细胞dim图 Rscript scripts/seurat_DimPlot.r -i 02.cluster/hcc.rds \ --reduction umap --group.by TissueTypeAbbr --pt.size 0.1 -o 02.cluster \ -p TissueType.umap #不同类型细胞dim图 Rscript scripts/seurat_DimPlot.r -i 02.cluster/hcc.rds \ --reduction umap --group.by HCC --pt.size 0.1 -o 02.cluster \ -p isHCC.umap #肿瘤和正常样本分开绘制 Rscript scripts/seurat_DimPlot.r -i 02.cluster/hcc.rds \ --reduction umap --group.by seurat_clusters --split.by HCC --pt.size 0.1 -o 02.cluster \ -p HCC.split.umap -W 15
聚类分析结果展示:
3.3 拟时序分析
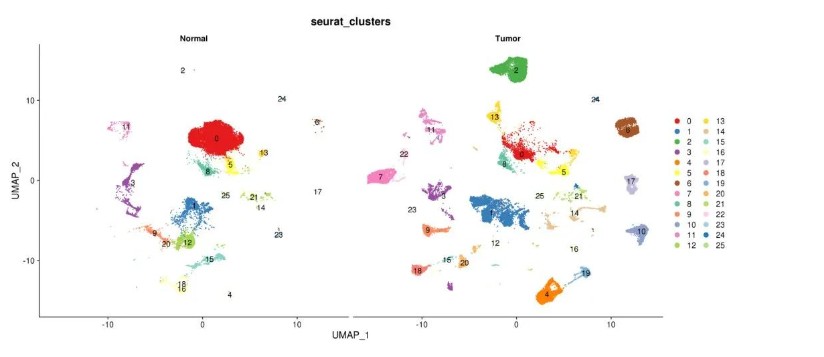
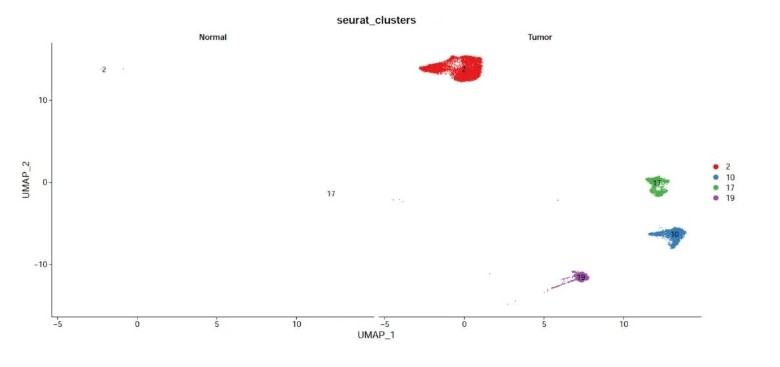
筛选出几团肿瘤样本特有的细胞亚群做拟时序分析,通过观察肿瘤样本和正常组织样本分开的散点图,进一步对比筛选其中的cluser2、17、10、19 来进行后续的拟时序分析。
筛选过程可以通过以下代码实现:
#4.1 筛选 肿瘤样本特有的细胞亚群 ###筛选方法1: Rscript scripts/subset_seurat_obj.r -i 02.cluster/hcc.rds \ --idents 2 17 10 19 \ -o 04.trajectory -p hcc.tumorSub #查看 筛选后的结果 Rscript scripts/seurat_DimPlot.r -i 04.trajectory/hcc.tumorSub.rds \ --reduction umap --group.by seurat_clusters --split.by HCC \ --pt.size 0.1 -o 02.cluster \ -o 04.trajectory -p HCC.sub.split.umap -W 15 --label
筛选到的细胞单独聚类分析之后发现,这些亚群明显不出现在normal组中:
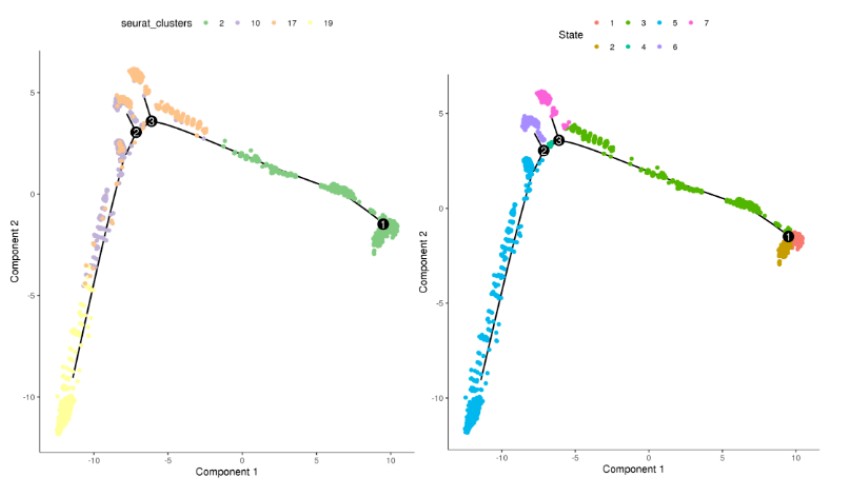
接下来就可以可利用monocle2做拟时序分析,具体代码如下所示:
##4.2 monocle2 拟时序分析 , 注意 约10000个细胞电脑内存至少大于 25G Rscript scripts/cell_trajectory.r \ -i 04.trajectory/hcc.tumorSub.rds -o 04.trajectory -p hcc.tumorSub
总结
利用代码命令可批量重复分析数据,这可以大大提高数据分析效率。虽然文章中的方法部分所展示的信息不是很全面,某些参数的设置并没有明确说明。但是,我们依旧可以利用自己撰写的代码复现文章的分析内容。只是在结果上和文章有细微差别,但这并不影响分析结果总体的准确性和一致性。
参考文献:
Li C, Chen J, Li Y, Wu B, Ye Z, Tian X, Wei Y, Hao Z, Pan Y, Zhou H, Yang K, Fu Z, Xu J, Lu Y. 6-Phosphogluconolactonase Promotes Hepatocellular Carcinogenesis by Activating Pentose Phosphate Pathway. Front Cell Dev Biol. 2021 Oct 26;9:753196. doi: 10.3389/fcell.2021.753196. PMID: 34765603; PMCID: PMC8576403.
Lu J, Chen Y, Zhang X, Guo J, Xu K, Li L. A novel prognostic model based on single-cell RNA sequencing data for hepatocellular carcinoma. Cancer Cell Int. 2022 Jan 25;22(1):38. doi: 10.1186/s12935-022-02469-2. PMID: 35078458; PMCID: PMC8787928.
- 发表于 2022-06-09 16:52
- 阅读 ( 6273 )
- 分类:转录组
