BUSCO评估基因组组装和完整性详解
BUSCO是Benchmarking Universal Single-Copy Orthologs(通用单拷贝同源基因基准)的缩写,基于基因进化(有参比对)评估基因组组装和注释完整性的开源python软件。
BUSCO软件介绍
BUSCO是一款使用python语言编写的对转录组和基因组组装质量进行评估的软件。在相近的物种之间总有一些保守的序列,而BUSCO就是使用这些保守序列与组装的结果进行比对,鉴定组装的结果是否包含这些序列,包含单条、多条还是部分或者不包含等等情况来给出结果。
BUSCO 软件根据OrthoDB 数据库,构建了几个大的进化分支的单拷贝基因集。将转录本拼接结果与该基因集进行比较,根据比对上的比例、完整性,来评价拼接结果的准确性和完整性。
下载与安装
BUSCO官网:https://busco.ezlab.org/。
BUSCO软件需要依赖以下3种工具:
Augustus
HMMER
Blast+
BUSCO软件安装不难,没有安装的可参考官网安装说明,小编这里就不过多介绍了。
软件使用
BUSCO软件有2种分析方法:auto-lineage 和 lineage_dataset。
auto-lineage:自动匹配进化分支
busco -m MODE -i INPUT -o OUTPUT --auto-lineage
busco -m MODE -i INPUT -o OUTPUT --auto-lineage-prok
# or ignoring eukaryotes to save runtime, if compatible with your experimental goal.
busco -m MODE -i INPUT -o OUTPUT --auto-lineage-euk
# or ignoring non-eukaryotes to save runtime, if compatible with your experimental goal.
-i:基因组组装文件;
-o:输出目录;
--auto-lineage:自动匹配进化分支;
--auto-lineage-prok:自动匹配真核生物进化分支;
--auto-lineage-euk:自动匹配原核生物进化分支。
lineage_dataset:指定进化分支
busco -i AF04-12.fna --lineage_dataset ~/database/BUSCO/eukaryota_odb10 --out output -m genome --offline
-i:基因组组装文件;
--lineage_dataset:指定进化分支数据库路径;
--out:输出目录;
-m :BUSCO分析模式,基因组组装;
--offline:关闭BUSCO下载文件。
要指定进化分支数据库路径就需要提前下载数据库,MANAUAL中提供了lineage数据源,数据库链接:https://busco-data.ezlab.org/v5/data/。
V5最新版的数据库:
需要下载并解压。
wget -c https://busco-data.ezlab.org/v5/data/lineages/bacteria_odb10.2020-03-06.tar.gz
tar -zxvf bacteria_odb10.2020-03-06.tar.gz
结果解读
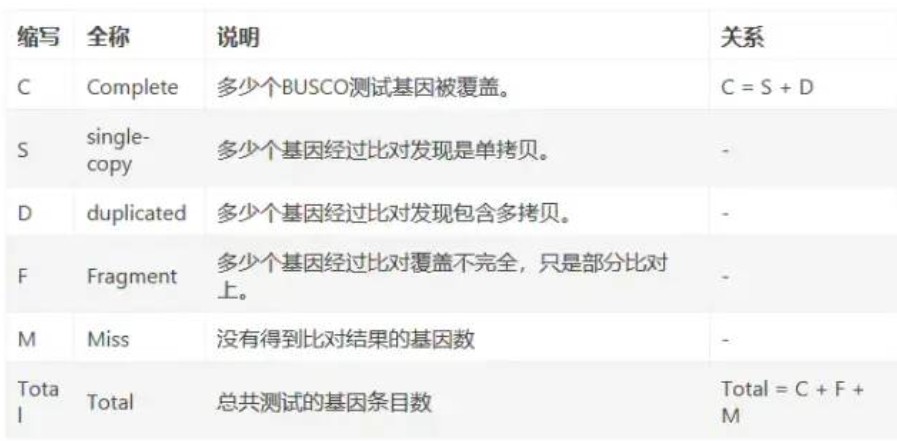
输出目录下的short_summary*.txt 文件为BUSCO评估结果文件。
格式如下:
# BUSCO version is: 5.3.2
# The lineage dataset is: eukaryota_odb10 (Creation date: 2020-09-10, number of genomes: 70, number of BUSCOs: 255)
# Summarized benchmarking in BUSCO notation for file /share/nas5/wangq/project/genome_assembly/liuch/ann/2.assemble/scaffolds.fasta
# BUSCO was run in mode: genome
# Gene predictor used: metaeuk
***** Results: *****
C:93.3%[S:92.5%,D:0.8%],F:1.2%,M:5.5%,n:255
238 Complete BUSCOs (C)
236 Complete and single-copy BUSCOs (S)
2 Complete and duplicated BUSCOs (D)
3 Fragmented BUSCOs (F)
14 Missing BUSCOs (M)
255 Total BUSCO groups searched
Dependencies and versions:
hmmsearch: 3.3
metaeuk: aa7ac2eb7334405ad57d50d78361e3dcd61bb27a
好啦,今天就先讲到这了,有兴趣的赶快试一下吧!
此外,我们在网易云课堂上有各种教学视频,有兴趣可以了解一下:
1. 文章越来越难发?是你没发现新思路,基因家族分析发2-4分文章简单快速,学习链接:基因家族分析实操课程
2. 转录组数据理解不深入?图表看不懂?点击链接学习深入解读数据结果文件,学习链接:转录组(有参)结果解读;转录组(无参)结果解读
3. 转录组数据深入挖掘技能-WGCNA,提升你的文章档次,学习链接:WGCNA-加权基因共表达网络分析
4. 转录组数据怎么挖掘?学习链接:转录组标准分析后的数据挖掘
6. 更多学习内容:linux、perl、R语言画图,更多免费课程请点击以下链接:
- 发表于 2022-09-09 14:01
- 阅读 ( 6837 )
- 分类:软件工具
