最全进化树构建知识点大盘点
系统进化树介绍
研究分子进化所要构建的系统发生树(Phylogenetic tree),也叫分子树,对于一个未知的基因或蛋白质序列,可以利用系统发生树确定与其亲缘关系最近的物种。比如你得到了一个新发现的细菌的核糖体RNA,你可以将它跟所有已知的核糖体RNA放在一起,然后用他们构建一棵系统发生树。这样就可以从树上推测出谁和这个新细菌的关系最近。系统发生树还可以预测一个新发现的基因或蛋白质的功能。系统发生树还分为有根树和无根树(下图),顾名思义,有根树就是有根,无根树就是无根。其实两者是可以互换的。如果我们按住无根树上某一个点,然后用把梳子将树上所有的枝条都以这个点为中心向右梳理,就能把它梳成有根树的样子。按住的这个点就是根。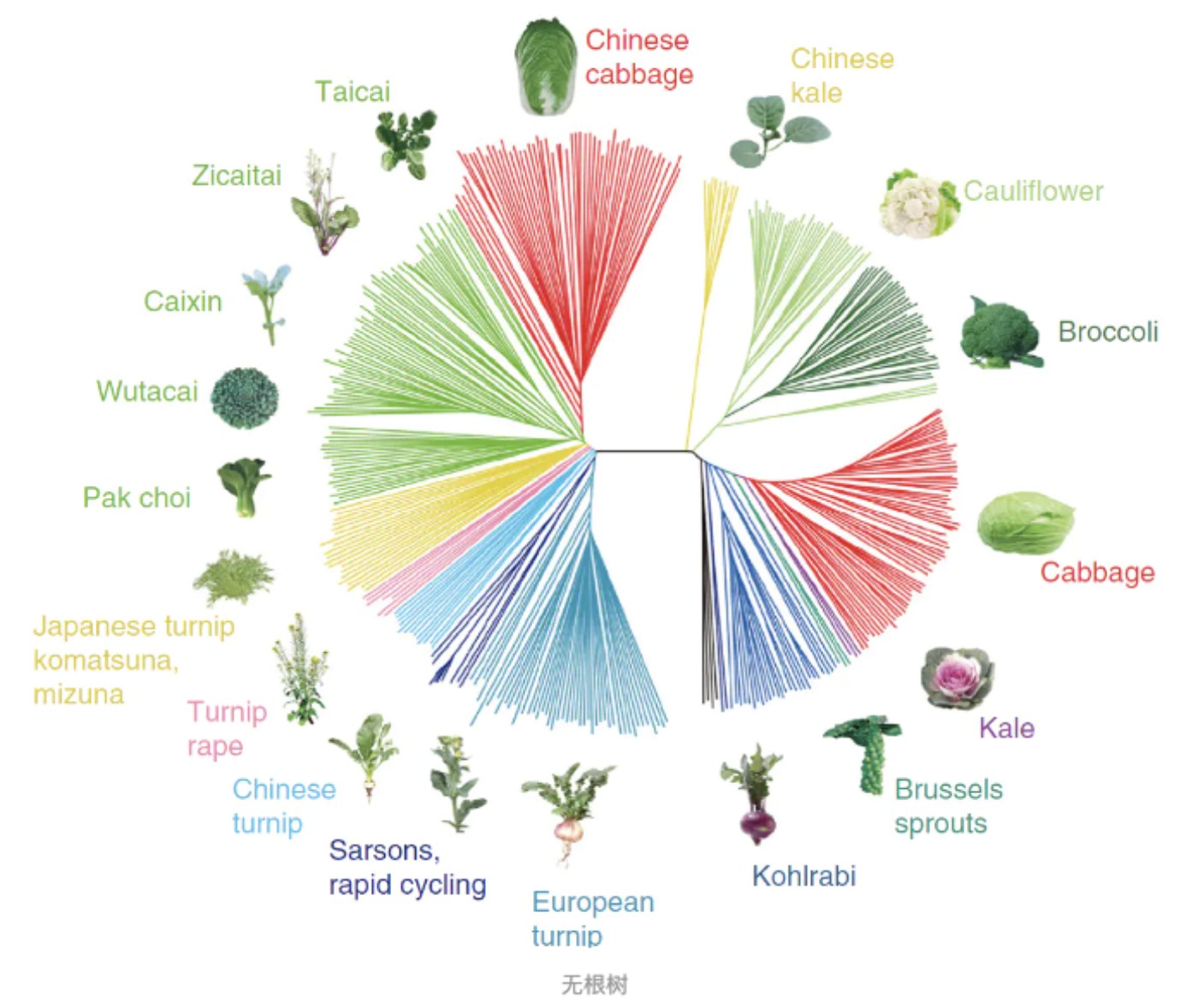 所以对于一棵树来说,根的位置是主观的,你想让他在哪它就在哪里。但是你不能随意指定哪个内节点当根,毕竟根有其自身的生物学意义,它应该是所有叶子的共同祖先。
所以对于一棵树来说,根的位置是主观的,你想让他在哪它就在哪里。但是你不能随意指定哪个内节点当根,毕竟根有其自身的生物学意义,它应该是所有叶子的共同祖先。
那么我们如何确定根的位置呢?可以通过外类群(outgroup)来确定,从而把无根树变成有根树。有根树反映了树上基因或蛋白质进化的时间顺序,通过分析有根树的树枝的长度,可以了解不同的基因或蛋白质以什么方式和速率进化。而无根树只反映分类单元之间的距离,而不涉及谁是谁的祖先问题。做有根树需要指定外类群。所谓外类群,就是你所研究的内容之外的一个群。比如你要分析某一个基因在不同人种间的进化关系,那就可以额外选择黑猩猩加入进来,作为外类群一同参与建树。
进化树的构建过程与方法
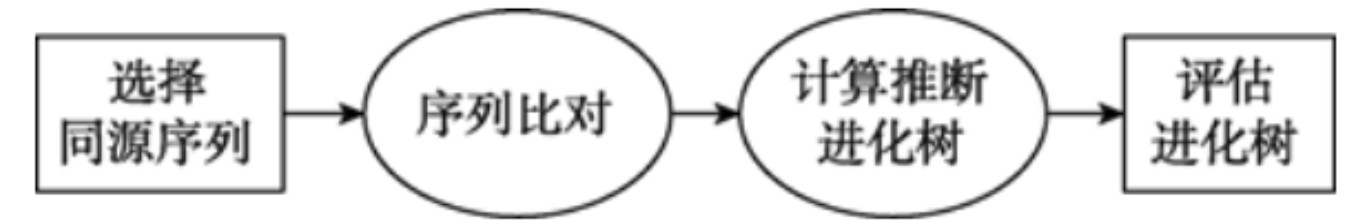
(1)数据准备:在进行系统发育分析时需要通过构建系统发育树来描述不同物种或者基因之间的进化关系,通过同源DNA的核苷酸序列或者同源蛋白质分子的氨基酸序列可以实现构建进化树的构建。
(2)序列比对:为了保证序列的同源性和所得系统发育关系的可靠性,需要对原始序列进行比对和校正。自动比对序列的软件包括Clustalw 、MAFFT、MUSCLE等。
(3)保守区:用于构建进化树的保守区选择是系统发育分析过程中一个重要的步骤。分析时可以选择保守位点,也可以选择基因全长序列,但是当序列差异大时,建议保留保守序列用于进化树构建。常用的保留序列保守区的软件有Gblock、trimAI等。
(4)进化树构建方法:
前期的序列处理好之后,我们基于序列间的差异就可以构建进化树了,构建进化树有很多方法:
① Distance-based methods 距离法:距离法的理论基础是最小进化原理(minimum evolution,ME),这类方法首先构造一个距离矩阵来表示每两个物种之间的进化距离,然后基于这个距离矩阵,采用聚类算法对研究的物种进行分类。通过不断的合并距离最小的两个节点和构建新的距离矩阵,最终得出进化树。
② Character-based methods 离散特征法:基于离散特征的系统发育树重构算法通过搜索各种可能的树,从中选出最能够解释物种之间进化关系的系统发育关系树,这类方法可以使用各种核酸和氨基酸替换模型。
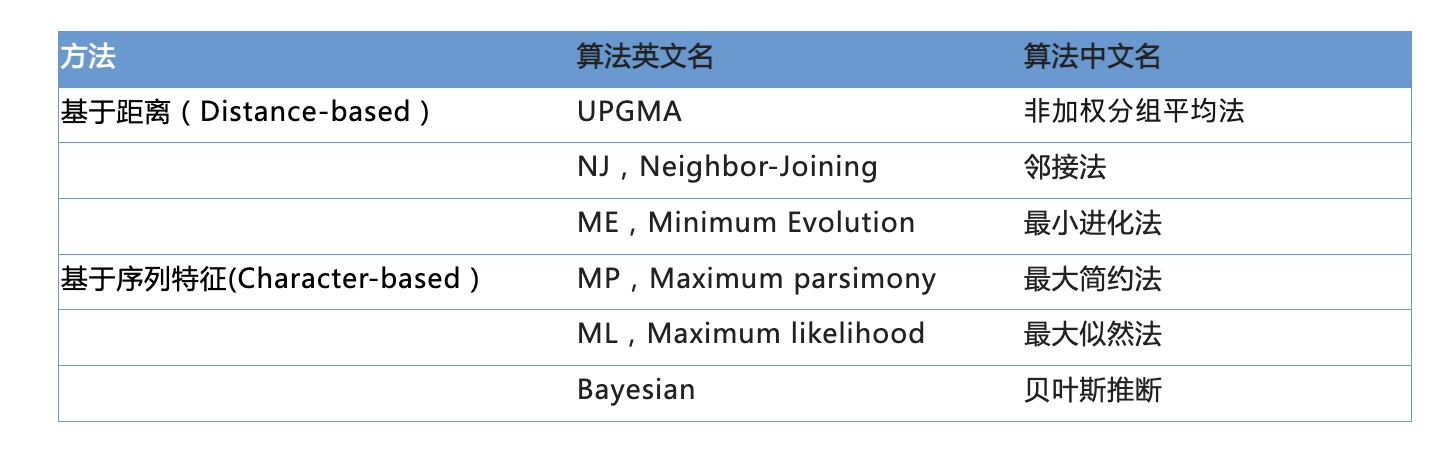
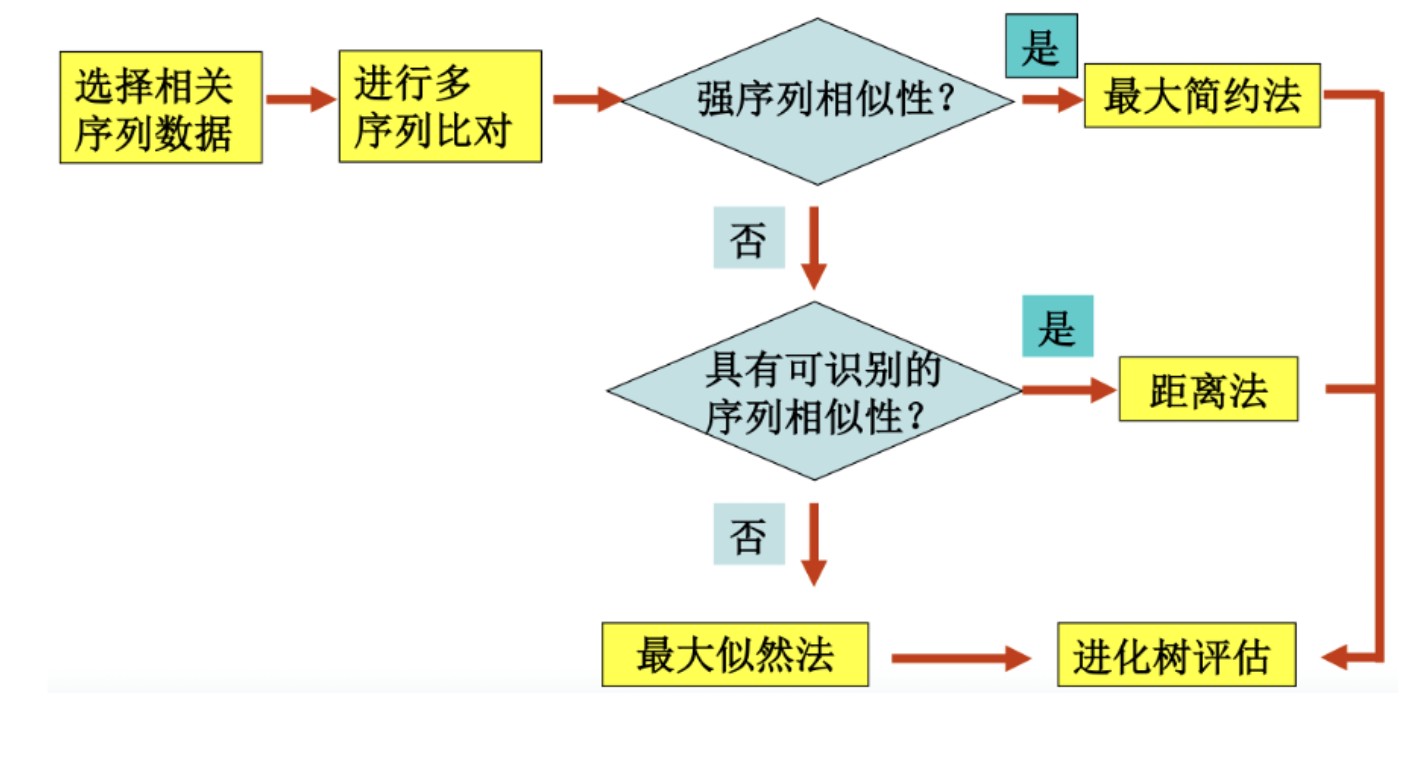
从计算速度来看,最快的是基于距离的方法,几十条序列几秒钟即可完成,其次是最大简约法,最大似然法就要慢得多,最慢的是贝叶斯法。但是从计算准确度来看,算得最慢的贝叶斯法确是最准确,而算得最快的基于距离法结果确是最粗糙。从实用的角度,建议使用最大似然法。因为这种方法无论从速度还是准确度都比较适中。
氨基酸/核酸替换模型的选择
要想构建一个最接近物种真实的进化关系的树,一般会选择基于序列特征的建树方法,通常是ML方法。这时,理解和选择合适的替换模型非常重要。选择替换模型是构建进化树的基础。这里从最简单的P距离模型说起:
p距离(p-distance ):两条蛋白质序列之间的氨基酸差异数为d, 序列的氨基酸数目均为n,则P距离:

以下为不同物种血红蛋白相对人类的P距离计算得到距离矩阵,就可以用于进化树构建了;
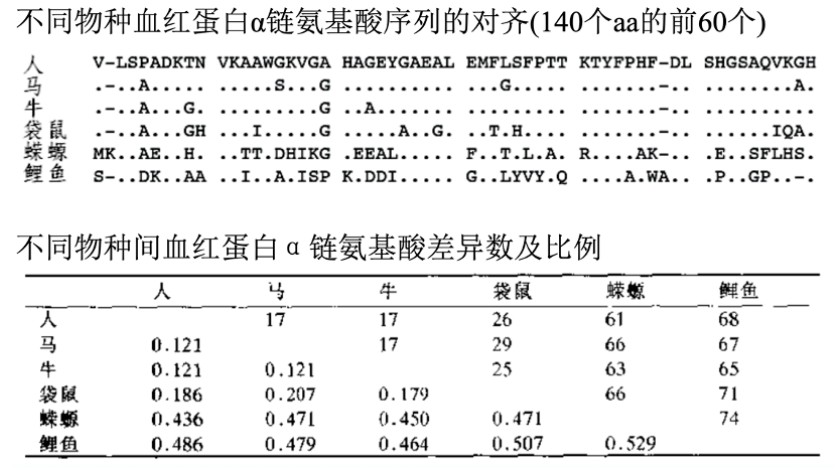
但是,p距离不是很严格地与进化时间成比例,进化时间较短时,两者大致成线性关系;当进化时间较长时,两者成非线性关系。
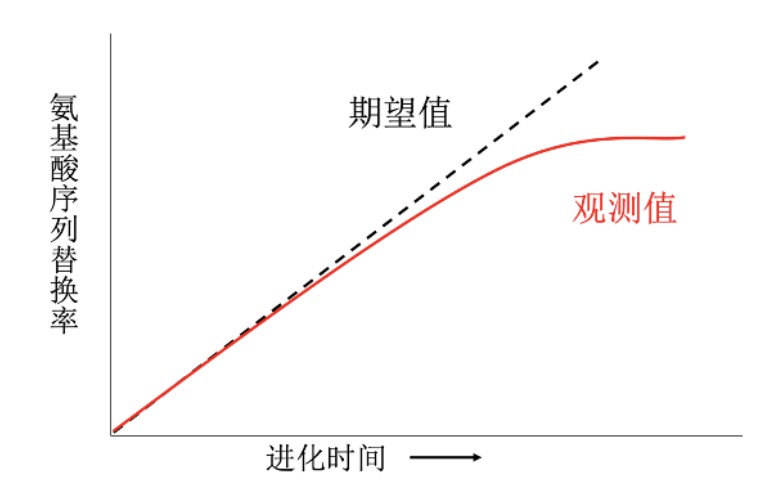
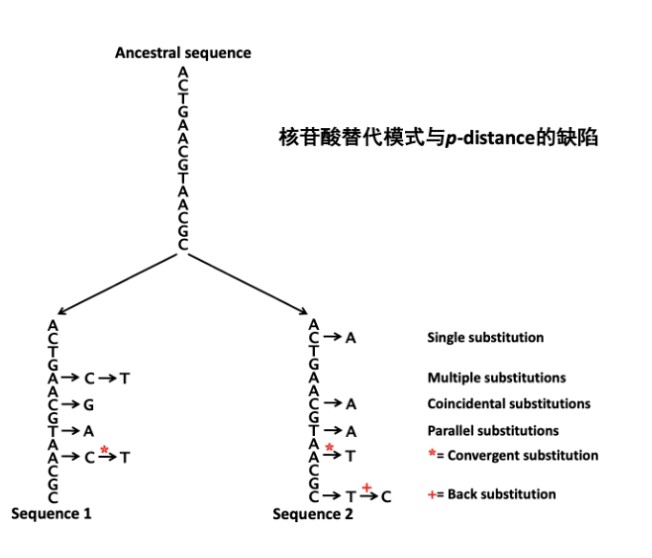
这是因为我们只能观察到现在的序列,祖先序列以及序列如何从祖先序列演化成现在序列的过程中发生了多少次替换我们并不知道。例如下图,祖先碱基C,通过回反替换C->T->C 又变成了C,我们观察到这个位点没有替换,但实际上已经发生了两次替换,这就是P距离的缺陷。
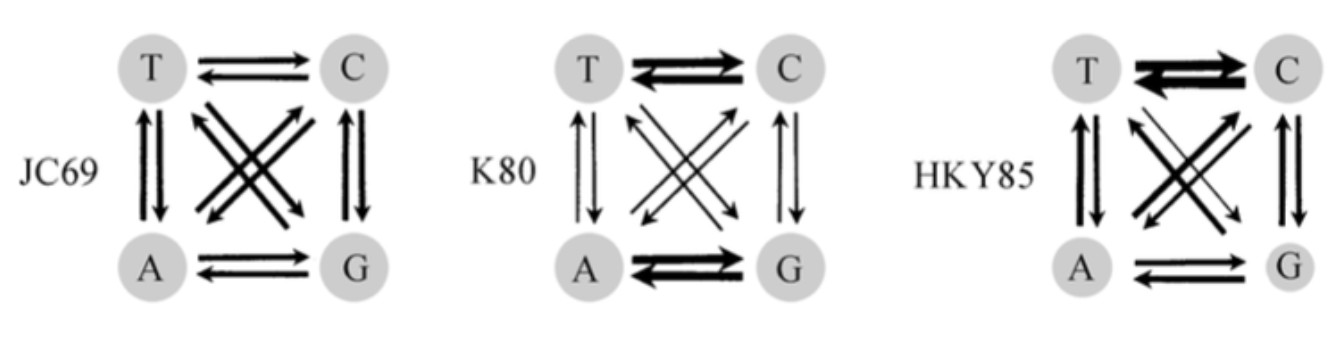
因此为解决p距离的问题,科学家们提出了很多替换模型,最简单的为JC69模型认为不同碱基之间的替换率相同(单参数模型);K80模型认为转换(A→G,G→A,C→T,T→C)和颠换(A→C,A→T,C→G,G→T)频率不同,认为转换频率比颠换频率高(双参数模型);HKY85更复杂, 又认为转换中的T→C的频率高于A→G的频率等。
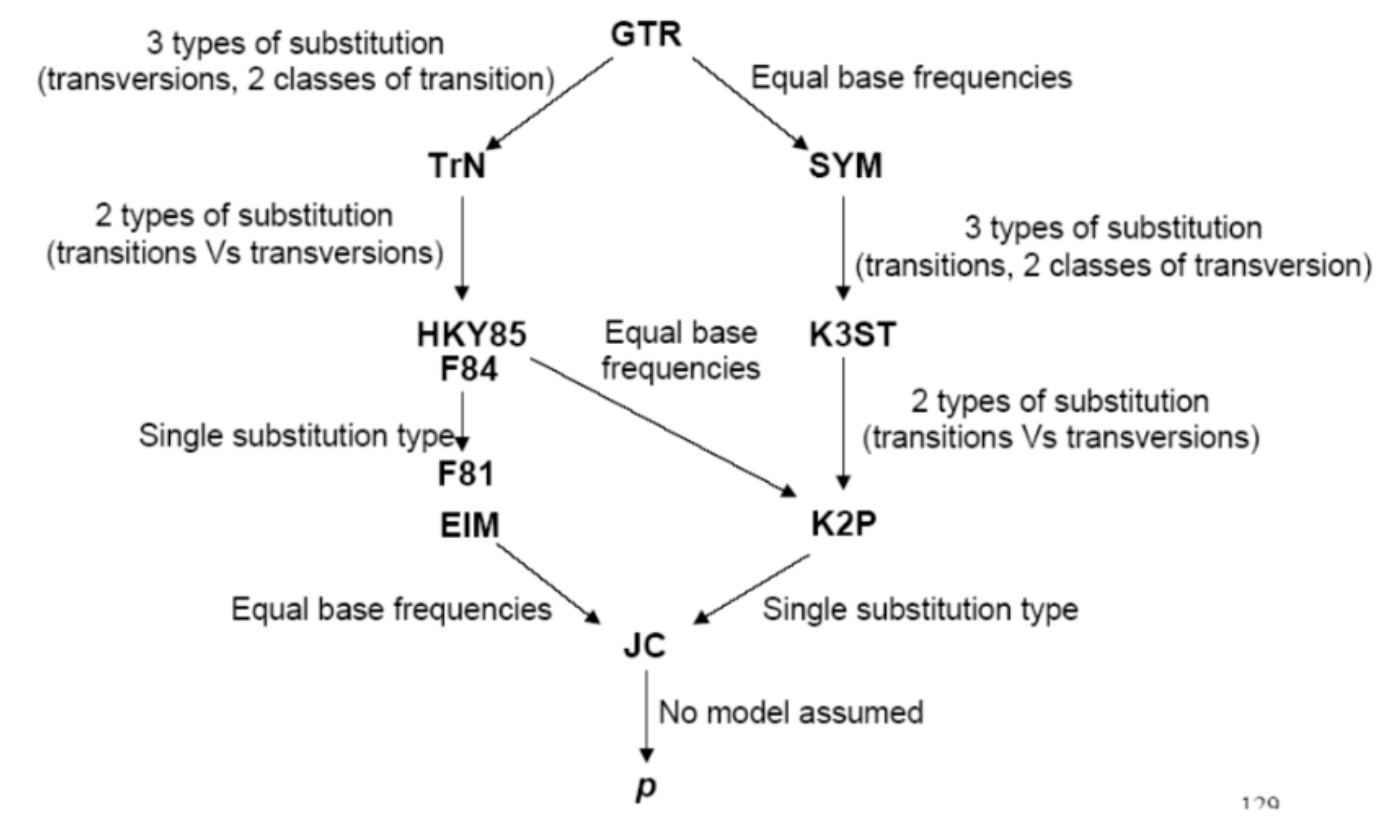
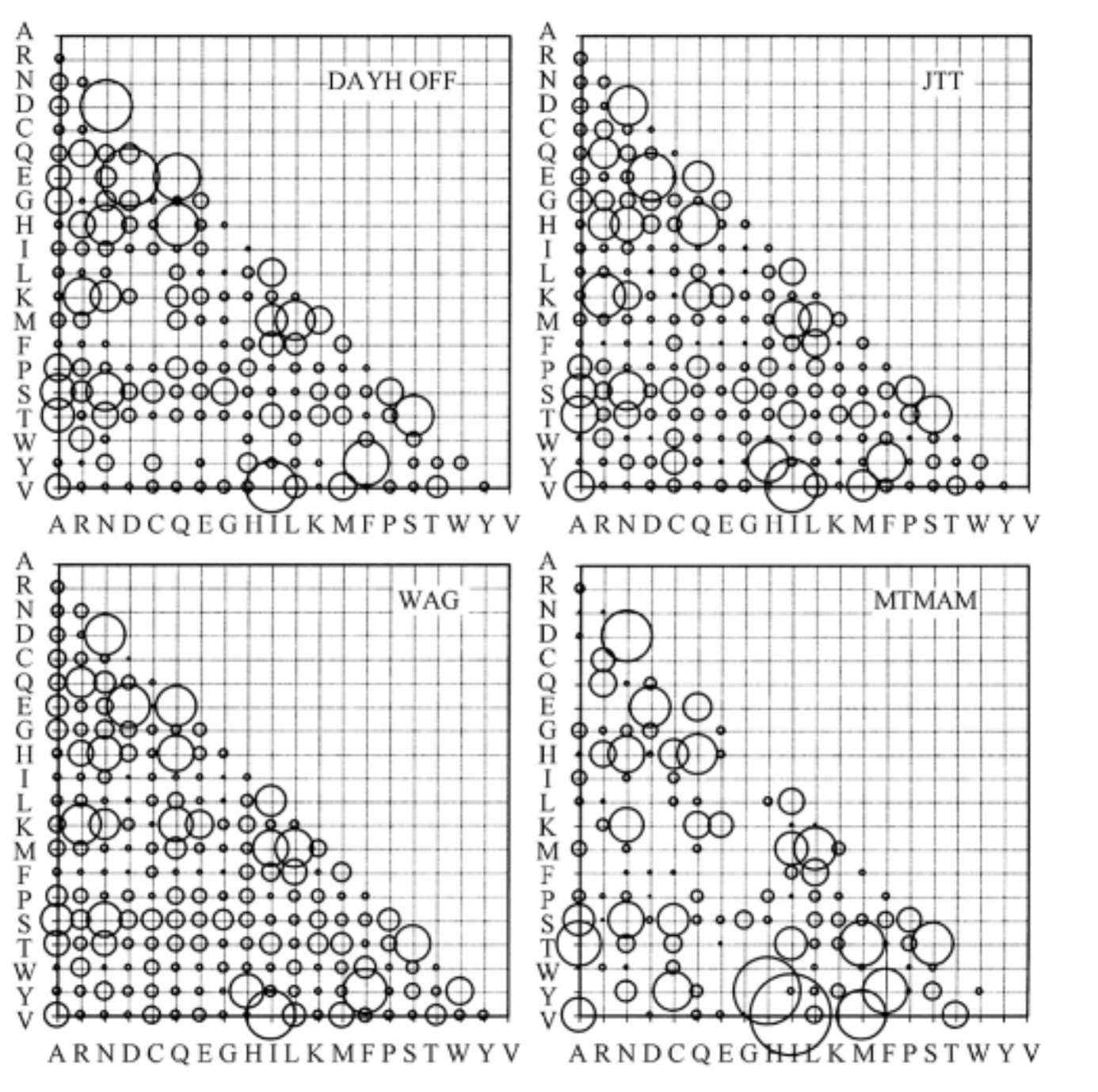
常用的核苷酸替换模型有JC69 、K80 、F81、TN93,HKY和GTR 模型等。其中GTR是所有模型中考虑参数最多最复杂的模型,其他所有模型是GTR模型的特例。上碱基的比例是不等的(两两之间),两两之间的替换速率也是不等的,所有这些参数都考虑进去了。
以上模型都是基于假设所有核苷酸位点的替代速率相同。例如:蛋白质的保守结构域所在的区域受到的选择压力大,进化速率慢相应的氨基酸的替代速率也慢,这是因为蛋白质不同区域在蛋白质结构与功能种所起的作用不同,因而所承受的选择压力不同,这种位点间的异质性可以用可变置换率表示,引入gamma分布(gamma distribution (G): gamma distributed rate variation among sites),gamma分布表示碱基/氨基酸位点之间的进化速率异质性
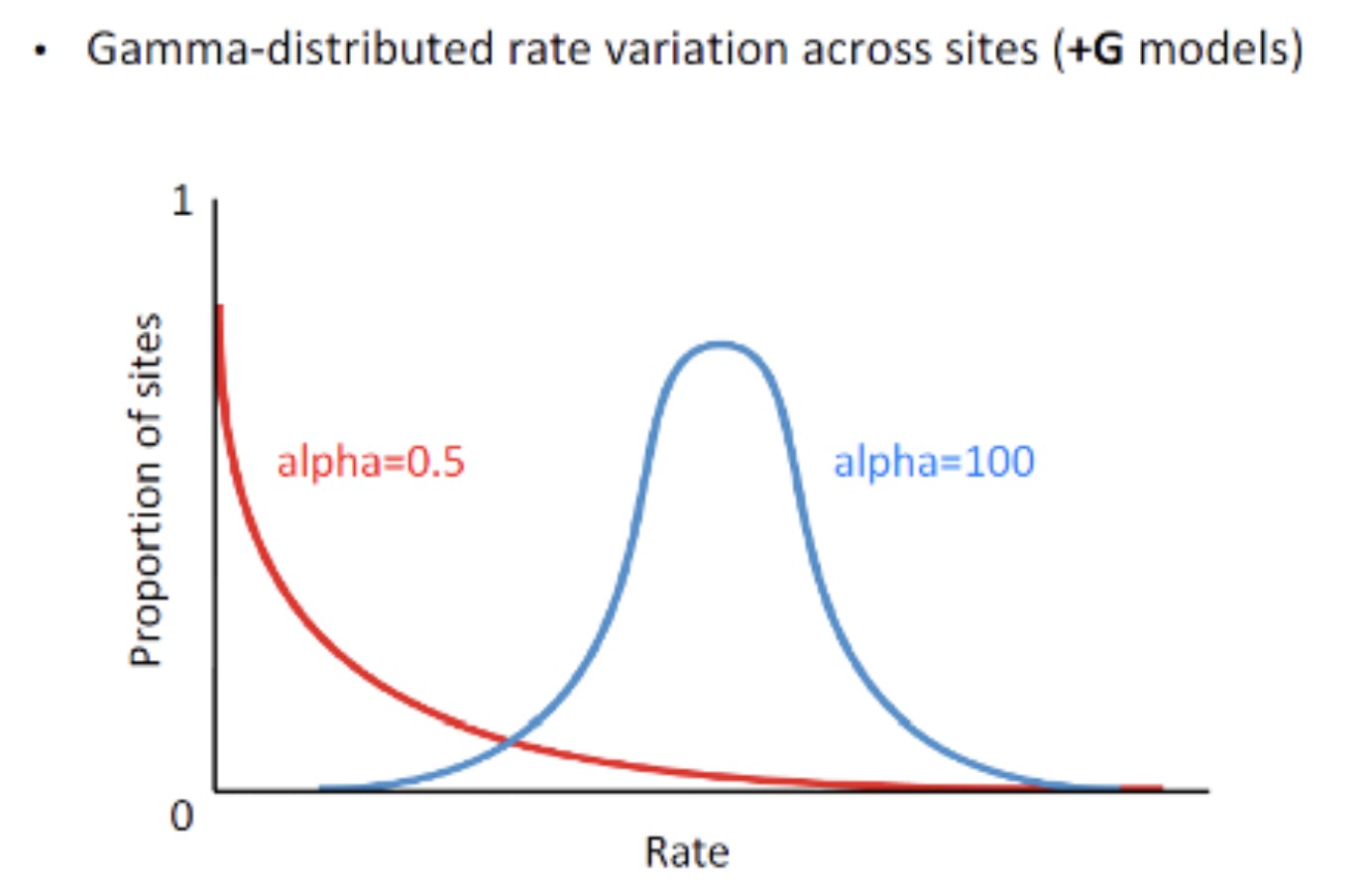
生物的进化历史是唯一的,我们并不能从这么多的模型中确定真实的核苷酸替代过程是依照哪种模型发生的。理论上应该尝试各种模型,根据检验结果选择最合适的模型进行计算。
在系统发育分析中,最大似然法(ML)和贝叶斯法(BI)是可以使用替换模型的两种算法,因此利用ML法或BI法重建系统发育树前,核苷酸替换模型的选择是必不可少的过程。
这里给出检验模型优劣的方法:似然率检验(Likelihood Ratio Test, LRT)用于比较两个模型对数据的拟合优度的一种统计检验方法;AIC值(Akaike Information criterion, AIC)是指Kullback-Leibler (K-L) 距离的估计值,K-L距离定义为一个模型被另一个模型替代后信息的丧失。因此,AIC值较小的模型更好;BIC(Bayesian Information Criterion, BIC):值越小,模型与数据拟合度越好;DT(Decision Tree,DT)将测量系统发生性能的相对分支长度误差整合到模型选择过程的方法。总结:BIC和DT具有较高的准确性和精确性,是目前最佳模型选择标准。对于核苷酸序列模型检验软件:ModelTest, JmodelTest, ModelFinder, Mega, ModelGenerator;对于氨基酸序列模型检验软件:ModelFinder, Protest, Mega, ModelGenerator。
进化树检验:Bootstrap
不同的方法可能会得到不同的结论,我们需要用不同的方法以及不同的参数,加上对生物问题的理解来构建最好的进化树来帮助我们更好的理解生物学问题。
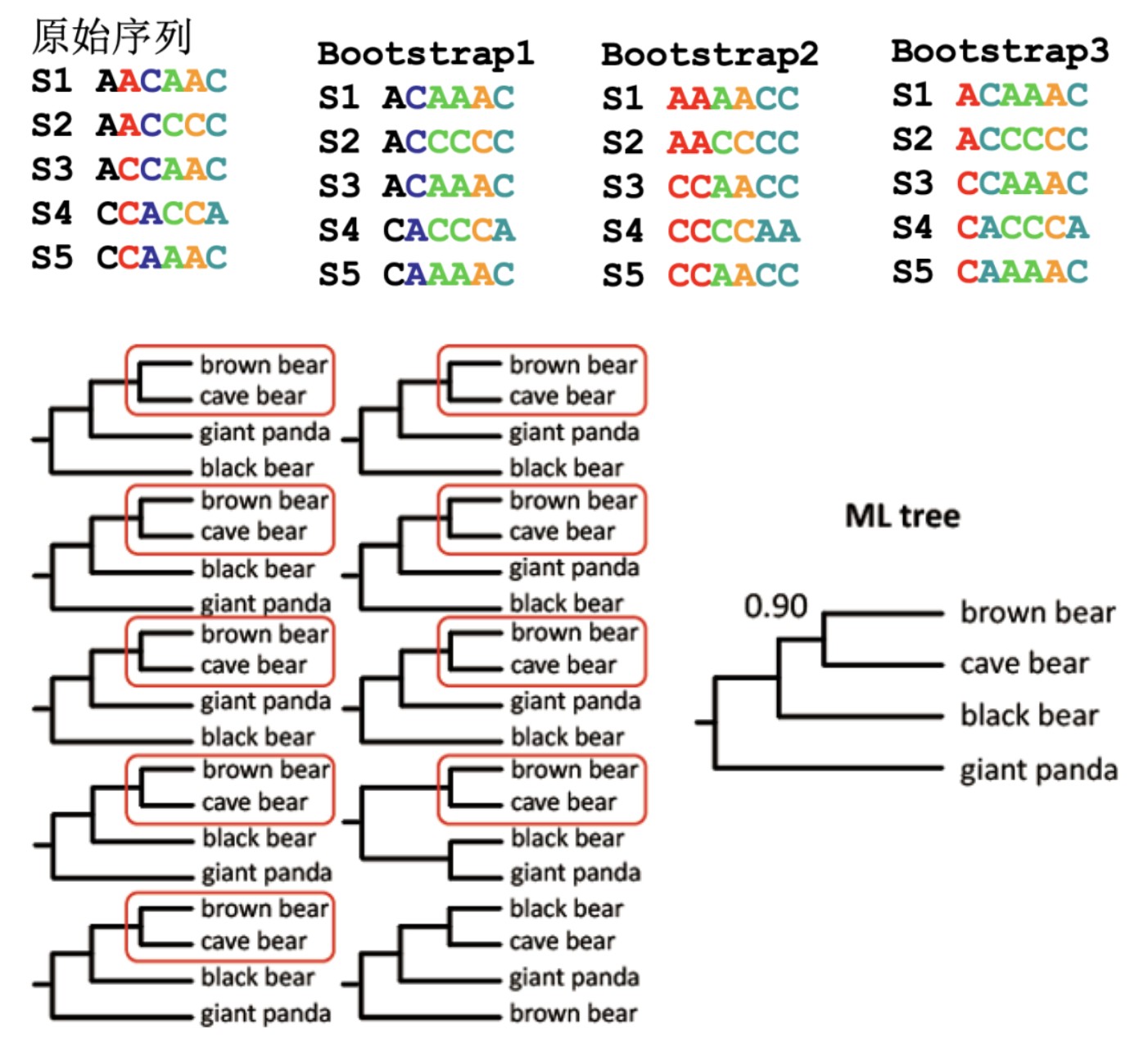
其中一个衡量树的好坏的方法就是看bootstrap的值,值越大越好。Bootstrap值是指根据所选的统计计算模型,设定初始值1000次,就是把序列的位点有放回的重抽样得到新的序列,再用相同的办法构树,如此让模型计算并绘制1000株系统发育树。如果原来树的分枝在后续构建的树中也出现了,就给这个分枝打上1分,如果没出现就给0分,这样给进化树打分后,每个分枝就都得出分值。系统发育树中每个节点上的数字则代表在1000次进化树分析中出现,有多少次最后一般换算成百分数。一般bootstrap的值>70%,则认为重建的进化树较为可靠。如果bootstrap的值太低,则有可能进化树的拓扑结构有错误,进化树是不可靠的。
因此,一般推荐用两种以上不同的方法构建进化树,如果所得到的进化树类似,且bootstrap值总体较高,则得到的结果较为可靠。通常情况下,只要选择了合适的方法和模型,构出的树均是有意义的,研究者可根据自己研究的需要选择最佳的树进行分析。
小结
比较以上几种主要的构树方法,一般情况下,若有合适的分子进化模型可供选择,用最大似然法构树获得的结果较好;对于近缘物种序列,通常情况下使用最大简约法,因为用的假设最少;而对于远缘物种序列,一般使用邻接法或最大似然法。
对于相似度很低的序列,邻接法往往出现长枝吸引(branch attraction)现象,有时严重干扰进化树的构建。对于各种方法重建进化树的准确性,Hall BG.( Mol Biol Evol 2005, 22(3):792-802)认为贝叶斯法最好,其次是最大似然法,然后是最大简约法。其实如果序列的相似性较高,各种方法都会得到不错的结果,模型间的差别也不大。再有贝叶斯的方法计算量巨大,速度太慢用的也不多。
- 发表于 2023-03-10 15:56
- 阅读 ( 13115 )
- 分类:基因组学
