dQTL.seq方法研究基因定位
dQTG.seq方法包含dQTG.seq1、dQTG.seq2、smoothLOD、C'、Δsnp index、ED六种计算方法。
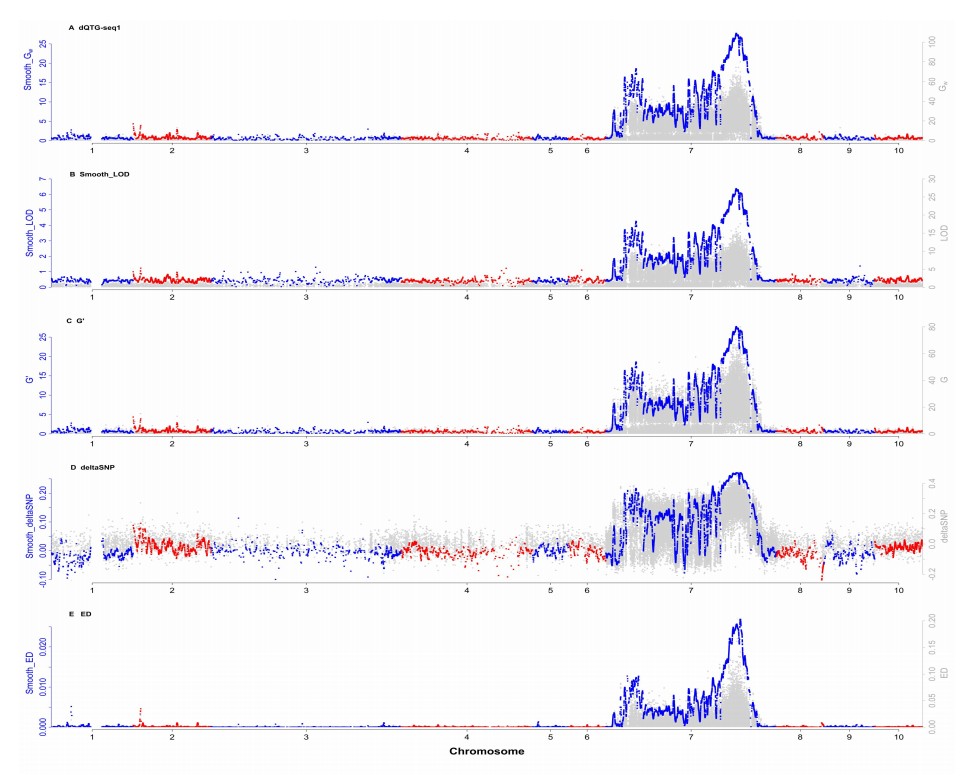
其中dQTG-seq1方法利用F2群体极端池标记等位基因数目来预测其基因型数目,通过观测的标记等位基因数目和预测的标记基因型数目构建了新统计量Gw,能显著提高基因检测功效。
其中dQTG-seq2方法可以处理基因效应较小或显性比太大的情况,对保留的每个极端表型个体DNA或RNA样品进行深度测序,利用观测的标记等位基因和基因型数目来检测小效应和极端超显性基因。
1. 工具准备
下载网址:https://cran.r-project.org/web/packages/dQTG.seq/index.html
install.packages("dQTG.seq") #下载dQTG.seq R包
library(dQTG.seq) #加载R包
QTG.seq((dir="/home", filegen="/home/BSA.csv", chr="all", color=c("blue","red"), CLO=NULL) #运行R包
dir:工作路径
filegen:输入文件
chr:结果中染色体选择,可以选择"all",计算并输出全部染色体;可以选择c(7,8),计算并输出7、8号染色体。
color:相邻染色体赋予不同颜色进行区分。
CLO:cpu数目。NULL表示默认,此时cpu为计算机上的cpu数量减去1,并且不超过10;自己设置时cpu数目最大也为10,如果超过10,默认为10。
2. 数据文件准备
01. 通过数据分析获得混池的snp位点信息all_sample_snp.vcf。
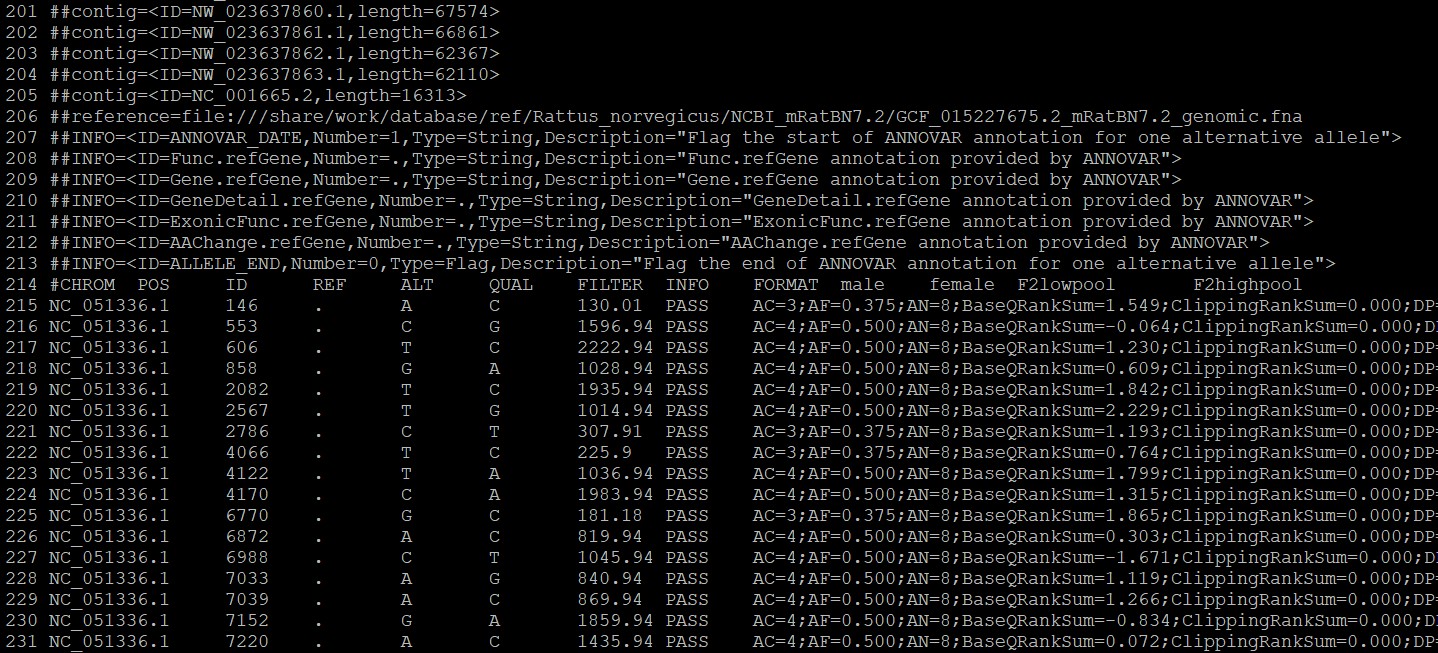 对all_sample_snp.vcf中的snp位点进行过滤,过滤掉亲本杂合以及亲本相同的位点。
对all_sample_snp.vcf中的snp位点进行过滤,过滤掉亲本杂合以及亲本相同的位点。
02. 格式转换
/share/work/biosoft/GATK/latest/gatk VariantsToTable -V all_sample_snp.vcf -F CHROM -F POS -F TYPE -GF AD -O output.table
利用VariantsToTable工具进行格式转换
-V:输入文件
-F:提取任一.vcf列,且每个字段将占据输出文件中的一列。这里命令行中只提取了CHROM、POS、TYPE三列。
-GF:后缀字段
-O:输出文件
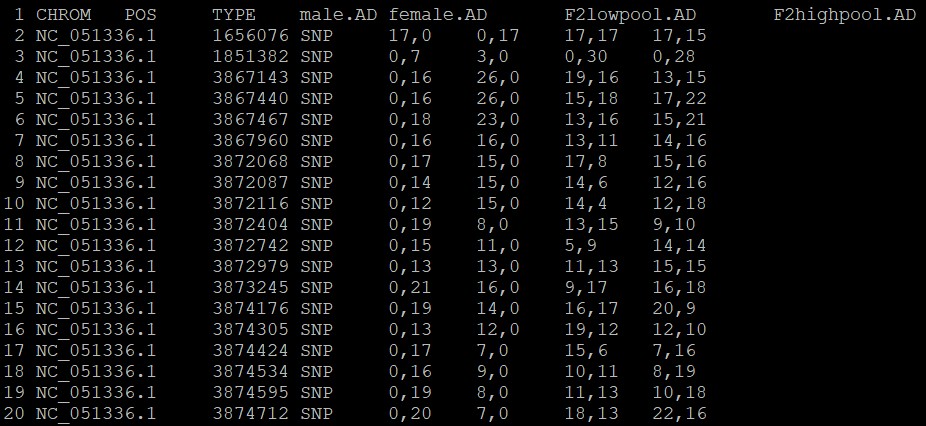 03. 数据整理。在dQTG.seq方法中亲本数据不是必须的。只提取子代极端混池中的数据
03. 数据整理。在dQTG.seq方法中亲本数据不是必须的。只提取子代极端混池中的数据
grep -v "CHROM" output.table|awk -F "\t|," '{print NR"\t"$1"\t"$2"\t"$8"\t"$9"\t"$10"\t"$11}' >output.table2
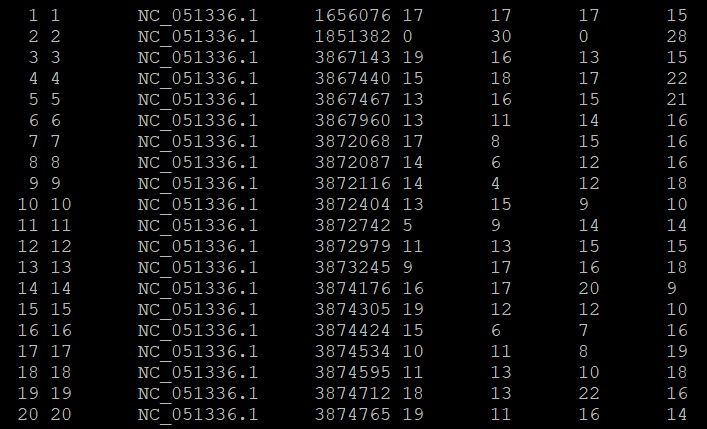 04. 添加表头信息,为了便于纠错将分隔符设置为分号
04. 添加表头信息,为了便于纠错将分隔符设置为分号
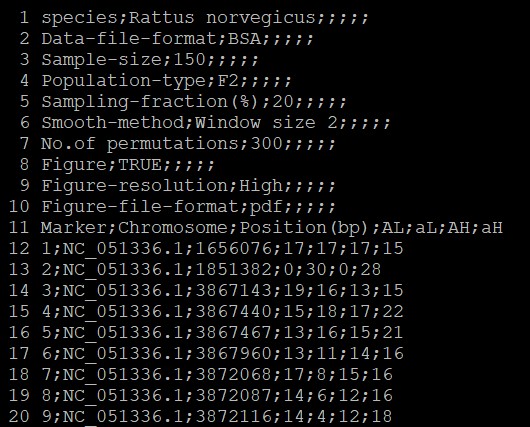 Species:物种名称
Species:物种名称
Data-file-format:文件格式,一共可识别五种文件格式:BSA,Extreme individuals,CIM,ICIM,GCIM。
Sample-size:子代混池中个体数目。The number of individuals in F2 population
Population-type:实验室中样本获取方式,可选择四种方式:F2, BC (backcross), DH (doubled haploid), and RIL (recombinant inbred line).
Sampling-fraction (%) :混池中极端个体所占比例
Smooth-method:平滑模式,可选择四种平滑模式:None,AIC,Window size,Block。默认为AIC。具体格式为“None”,"AIC",“Window size 0.5”或者”Block 30”。另外Window size取值范围是0.5-30。
No. of permutations:确定有效QTN阈值时的排列实验次数,默认为300。
Figure:是否生成图片。Figure=False: no figure output; Figure=True: the output of figures from different methods. The default is True.
Figure-resolution:图片分辨率。Figure-resolution=Low: the figure with low resolution; Figure-resolution=High: the figure with high resolution. The default is High.
Figure-file-format:图片格式。Figure-file-format= jpeg, png, tiff, or pdf. jpeg indicates the *.jpeg format of figure file. Please see Table 4.
第11行为列名,选择示例数据列名。其中后四列“AL\taL\tAH\taH”。代表分别在低(L)和高(H)池中的等位基因A和A的数量。
3. 运行dQTG.seq包
library(dQTG.seq)
file <- read.table("output.table2",sep = ";",fill=T)
dQTG.seq(dir="./",filegen=file,chr="all",color=c("blue","red"),CLO=10)
4. 结果
- 发表于 2023-03-29 11:56
- 阅读 ( 3190 )
- 分类:R
