bam文件格式详解
sam是短序列比对默认的标准格式,是以TAB为分割符的文本格式。主要应用于测序序列mapping到基因组上的结果表示,另外也可以表示其他的多重比对结果。一般把测序reads比对到参考基因组以后,通常得到的就是sam文件。BAM就是SAM的二进制文件,具有更小的存储空间,并且许多下游分析工具使用的是BAM格式。使用samtools view xxx.bam | less -SN 可以查看二进制的bam文件。
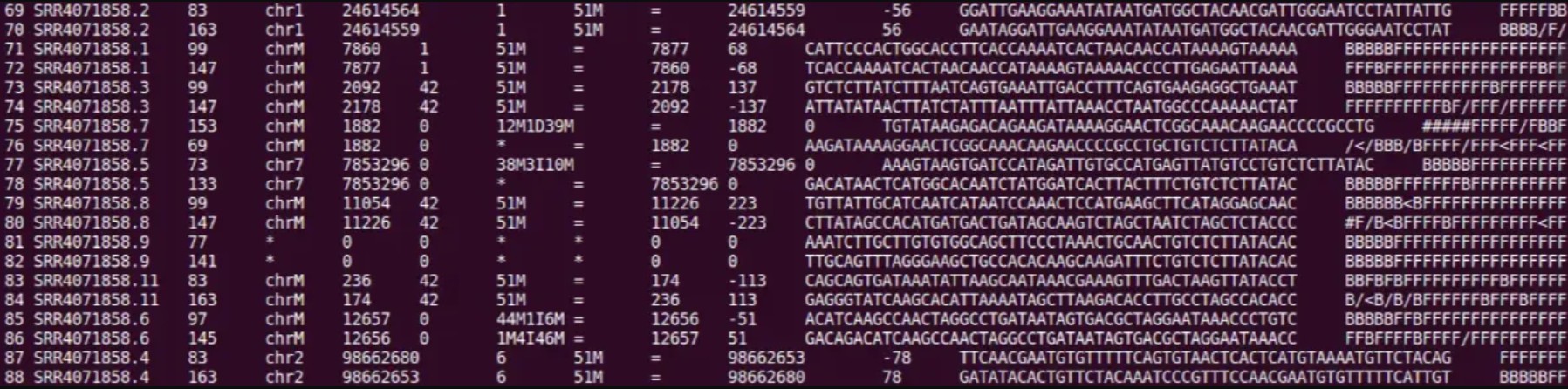
第1列:fastq的read ID
第2列:FLAG(如果某一个数值不是下面的任意值,那么那个数值就是下面这些数里面几个的和)。另外,如果flag值是0,那么说明测序为单端测序且这条read是primary line,一般是该read的最佳比对。
1:该read是成对的paired reads中的一个
2:paired reads中每个都正确比对到参考序列上
4:该read没比对到参考序列上
8:与该read成对的matepair read没有比对到参考序列上
16:该read其反向互补序列能够比对到参考序列
32:与该read成对的matepair read其反向互补序列能够比对到参考序列
64:在paired reads中,该read是与参考序列比对的第一条
128:在paired reads中,该read是与参考序列比对的第二条
256:该read是次优的比对结果
512:该read没有通过质量控制
1024:由于PCR或测序错误产生的重复reads
2048:补充匹配的read
只有一条reads没有比对上:73, 133, 89, 121, 165, 181, 101, 117, 153, 185, 69, 137
两条reads都没有比对上:77、141
比对上了,方向也对,并且在插入片段大小范围内:99, 147, 83, 163
比对上了,也在插入片段大小范围内, 但是方向不对:67, 131, 115, 179
唯一配对,就是插入片段大小范围不对:81, 161, 97, 145, 65, 129, 113, 177
这里说一下secondary alignment(256)和supplementary alignment(2048)的区别,256代表的是该条read比对到多个位置,该条记录为次优比对,在双端测序中,代表hardclip,而2048标记为该条比对记录为该read的补充比对记录,就是嵌合比对,这些记录是相互的补充,有可能只是这条read的一部分,在二代测序的结果中很容易找到很多的hardclip记录,但是2048的记录很少,在三代测序的比对结果中常见2048的supplementary alignment。
使用samtools view -f/-F参数可以获取包含/不包含指定flag的内容的条目。
第3列:染色体名称。如果这列是“ * ”,可以认为这条read没有比对上的序列,则这一行的第四,五,八,九 列是“0”,第六,七列与该列是相同的表示方法。
第4列:比对的位置,从对应上的染色体第1位开始往后计算。没有比对上的,此处为0。
第5列:MAPQ比对质量值。越高说明该read比对到参考基因组上的位置越唯一,0表示在参考基因组有多种定位的可能性。60表示在参考基因组只有这一种定位位置。
第6列: M表示匹配、I表示插入、D表示删除、N表示内含子和D类似、S表示替换、H表示剪切。
比如3S6M1P1I4M,前三个碱基被剪切去除了,然后6个比对上了,然后打开了一个缺口,有一个碱基插入,最后是4个比对上了,是按照顺序的;
比如:36M 表示36个碱基在比对时完全匹配。
比如:如37M1D2M1I,这段字符的意思是37个匹配,1个参考序列上的删除,2个匹配,1个参考序列上的插入。
(clipped均表示一条read的序列被分开,之所以被分开,是因为read的一部分序列能匹配到第三列的RNAME序列上,而被分开的那部分不能匹配到RNAME序列上。而H只出现在一条read的前端或末端,但不会出现在中间,S一般会和H成对出现,当有H出现时,一定会有一个与之对应的S出现)
①S: 这部分没比对上但保留在了SAM/BAM比对结果中。
②H: 这部分没比对上并且没有保留在SAM/BAM比对结果中。
第7列: 这条reads第二次比对的位置。=表示参考序列与reads一模一样,*表示没有完全一模一样的参考序列。
第8列: 该列表示与该reads对应的mate pair reads的比对位置(即mate),若无mate,则为0。
第9列: 序列模板长度,如果同一个片段都比对上了同一个参考序列,为最左边的碱基位置到最右边的碱基位置(左为正,右为负)。当mate 序列位于本序列上游时该值为负值。不可用时,为0。
第10列: read的序列。
第11列: ASCII码格式的序列质量。格式同FASTQ一样。其中1、10、11合起来就是fq格式文件。
第12列: 可选的区域。格式类似AS:i:-1 XN:i:0 XM:i:1 XO:i:0 XG:i:0 NM:i:1 MD:Z:35T0 YT:Z:UU
AS:i 匹配的得分
XS:i 第二好的匹配的得分
YS:i mate 序列匹配的得分
XN:i 在参考序列上模糊碱基的个数
XM:i 错配的个数
XO:i gap open的个数
XG:i gap 延伸的个数
NM:i 经过编辑的序列
YF:i 说明为什么这个序列被过滤的字符串
YT:Z 值为UU表示不是pair中一部分(单末端?)、CP(是pair且可以完美匹配)
DP(是pair但不能很好的匹配)、UP(是pair但是无法比对到参考序列上)
MD:Z 代表序列和参考序列错配的字符串
线性对齐:一条read比对到参考序列上,可以存在插入(insert)、缺失(delete)、跳跃(skip)、剪切(clip),但是方向不变(不能是一部分和正链匹配,另一部分又和负链匹配),sam文件中只占用一行记录。
嵌合比对:由于一条测序read比对到基因组上时分别比对到两个不同的区域,而这两个区域基本没有接触和重叠。因此它在sam文件中需要占用多行记录显示。只有第一个记录称作"representative",其他的都是"supplementary"。RNA-seq中的chimeric read或许可以说明有融合基因存在,但在基因组中一般作为结构变异的证据。
链接:https://www.jianshu.com/p/50be38f6cbb8
https://blog.csdn.net/qq_35696312/article/details/101760397
- 发表于 2023-05-16 10:00
- 阅读 ( 8416 )
- 分类:基础知识
