rMATS turbo使用
rMATS turbo使用
rMATS(reproducible RNA-seq Analysis of Transcript Splicing)是一个用于RNA测序数据分析的工具,用于检测基因的剪接事件。rMATS Turbo 是 rMATS 的改进版本,专注于更高的性能和更快的速度,能够允许不同长度的reads进行分析。
rMATS可识别的可变剪切事件有5种:
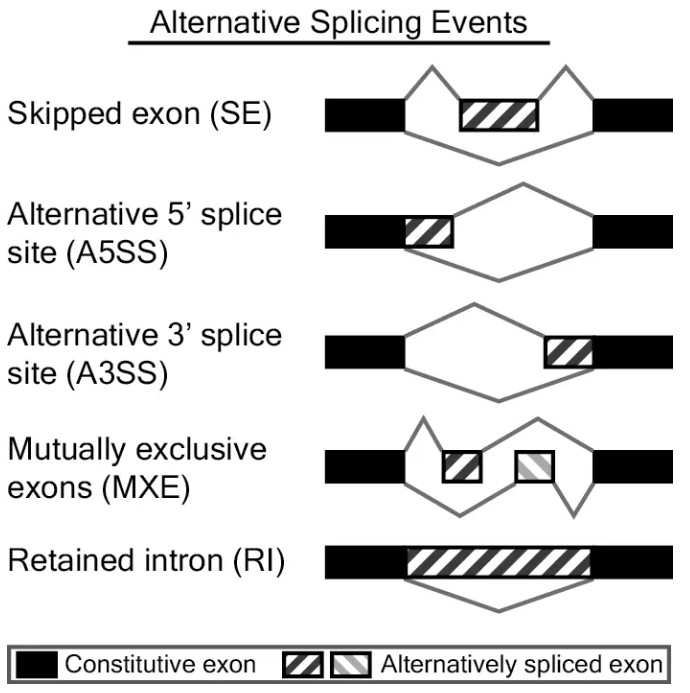
skipped exon (SE),外显子跳跃,指一个或多个外显子连同其两端的内含子一起被剪切,在成熟mRNA中不存在。
alternative 5' splice site (A5SS),5’端可变剪接,它们的3’端剪接位点一致但5’端剪接位点不同,产生不同长度的5’端外显子。
alternative 5' splice site (A3SS),3’端可变剪接,它们的5’端剪接位点一致但3’端剪接位点不同,产生不同长度的3’端外显子。
mutually exclusive exons (MXE),外显子互斥,成熟的mRNA变体中,彼此特有的外显子,这些外显子不能同时出现在同一成熟mRNA中。
retained intron (RI),内含子保留,在一些转录本中内含子不会被剪切掉,保留在成熟的mRNA。
定量
rMATS采用exon inclusion level 来定义样本中可变剪切事件的表达量,以外显子跳跃(Skipped Exon)为例,正常的转录本称之为Exon Inclusion Isofrom, 发生了外显子跳跃的转录本则称之为Exon Skipping Isofrom。
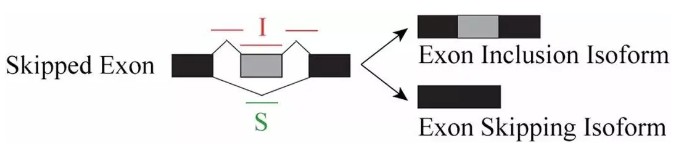
用 I 表示比对到Exon Inclusion Isofrom上的reads,S表示比对到Exon Skipping Isofrom上的reads, 则该外显子跳跃的可变剪切事件比例可以表示为:
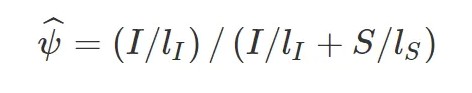
可以看到,exon inclusion level实际上是inclusion isofrom所占的比例,计算时,用长度校正了原始的reads数。其他类型的可变剪切事件也可以划分成上述两种isoform, 示意图如下:
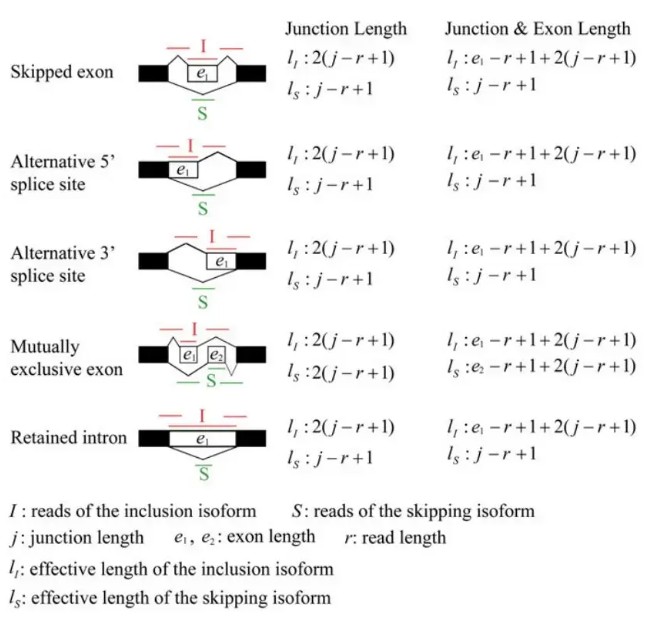
可以看到,rmats在计算isofrom的长度时,提供了两种方式,二者的区别就在于是否考虑跳过的exon的长度。
软件安装
conda create -n my_rmats_env conda activate my_rmats_env conda install rmats-turbo rmats-turbo --version
软件使用
参数说明:
python rmats.py -h
usage: rmats.py [options]
optional arguments:
-h, --help show this help message and exit
--version show program's version number and exit
--gtf GTF An annotation of genes and transcripts in GTF format
--b1 B1 A text file containing a comma separated list of the
BAM files for sample_1. (Only if using BAM)
--b2 B2 A text file containing a comma separated list of the
BAM files for sample_2. (Only if using BAM)
--s1 S1 A text file containing a comma separated list of the
FASTQ files for sample_1. If using paired reads the
format is ":" to separate pairs and "," to separate
replicates. (Only if using fastq)
--s2 S2 A text file containing a comma separated list of the
FASTQ files for sample_2. If using paired reads the
format is ":" to separate pairs and "," to separate
replicates. (Only if using fastq)
--od OD The directory for final output
--tmp TMP The directory for intermediate output such as ".rmats"
files from the prep step
-t {paired,single} Type of read used in the analysis: either "paired" for
paired-end data or "single" for single-end data.
Default: paired
--libType {fr-unstranded,fr-firststrand,fr-secondstrand}
Library type. Use fr-firststrand or fr-secondstrand
for strand-specific data. Default: fr-unstranded
--readLength READLENGTH
The length of each read
--variable-read-length
Allow reads with lengths that differ from --readLength
to be processed. --readLength will still be used to
determine IncFormLen and SkipFormLen
--anchorLength ANCHORLENGTH
The anchor length. Default is 1
--tophatAnchor TOPHATANCHOR
The "anchor length" or "overhang length" used in the
aligner. At least "anchor length" NT must be mapped to
each end of a given junction. The default is 6. (Only
if using fastq)
--bi BINDEX The directory name of the STAR binary indices (name of
the directory that contains the SA file). (Only if
using fastq)
--nthread NTHREAD The number of threads. The optimal number of threads
should be equal to the number of CPU cores. Default: 1
--tstat TSTAT The number of threads for the statistical model.
Default: 1
--cstat CSTAT The cutoff splicing difference. The cutoff used in the
null hypothesis test for differential splicing. The
default is 0.0001 for 0.01% difference. Valid: 0 <=
cutoff < 1. Does not apply to the paired stats model
--task {prep,post,both,inte}
Specify which step(s) of rMATS to run. Default: both.
prep: preprocess BAMs and generate a .rmats file.
post: load .rmats file(s) into memory, detect and
count alternative splicing events, and calculate P
value (if not --statoff). both: prep + post. inte
(integrity): check that the BAM filenames recorded by
the prep task(s) match the BAM filenames for the
current command line
--statoff Skip the statistical analysis
--paired-stats Use the paired stats model
--novelSS Enable detection of novel splice sites (unannotated
splice sites). Default is no detection of novel splice
sites
--mil MIL Minimum Intron Length. Only impacts --novelSS
behavior. Default: 50
--mel MEL Maximum Exon Length. Only impacts --novelSS behavior.
Default: 500
运行:
##/path/to/b1.txt /path/to/1_1.bam,/path/to/1_2.bam ##/path/to/b2.txt /path/to/2_1.bam,/path/to/2_2.bam python rmats.py --b1 /path/to/b1.txt --b2 /path/to/b2.txt --gtf Gallus_gallus.GRCg6a.101.gtf --od A_vs_B --tmp A_vs_B/tmp -t paired --variable-read-length --readLength 150 --cstat 0.0001 --libType fr-unstranded --novelSS --nthread 4
--b1 为组别1的bam文件的路径,若有生物学重复则bam文件路径用逗号隔开;
--b2 为组别2的bam文件的路径,若有生物学重复则bam文件路径用逗号隔开;
--gtf 为已知的基因及转录本的gtf文件;
--od 即为输出路径;
-t 测序类型为单端或者双端 ;
--variable-read-length 能够允许不同长度的长度的reads进行分析;
--readLength 若长度不一致时,可使用该参数将reads截取到给定的数值;
--libType 文库类型,可选择是否为链特异性;
--tmp 暂存目录;
--nthread 线程数。
输出文件详情请查看:https://github.com/Xinglab/rmats-turbo/blob/v4.2.0/README.md
- 发表于 2024-01-17 11:30
- 阅读 ( 1936 )
- 分类:软件工具
