你了解SAM和BAM文件吗
当我们测序得到的fastq数据map到基因组之后,会得到一个以sam或bam为扩展名的文件。这里,SAM的全称是sequence alignment/map format。而BAM就是SAM的二进制文件,也就是压缩格式的sam文件。 那么SAM文件的格式是什么样子的呢?这里给大家简单解释一下。
SAM格式简介
SAM文件由头文件和map结果组成。头文件为注释信息,以@开头,可有可无,就不做多介绍了。重要的是比对结果,例如这样的:
E00514:173:H3C3JCCXY:4:1124:12398:67234 337 Chr00 32904 0 150M Chr09 33498107 0 TCAATTTCACTTGAAGCTTACTTGTAGTTTCAGGCTTGGTCAAGCGCGATACAAACCATGTAGTAGGAGTCCTCCAAGTCGCCAAGCTAGGGGATCTGCTGAAAGAGGTGACAGACAAGGTAAGCAATCAGAGCTCTAAGCAATCAGTCC iieiiiii`eiiiiiiiiiiiiiiieiiiiiiiieiiiiiiiiiiiiiiiiiiiiieiiiiiiiiiiiiieiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiieiiiiiiiii`iiieeieieeieee`` AS:i:-6 XN:i:0 XM:i:1 XO:i:0 XG:i:0 NM:i:1 MD:Z:136C13 YT:Z:UU NH:i:8 CC:Z:Chr10 CP:i:18604313 HI:i:0 RG:Z:J36CK1
E00514:173:H3C3JCCXY:4:1124:12398:67234 369 Chr00 32904 0 150M Chr16 2469225 0 TCAATTTCACTTGAAGCTTACTTGTAGTTTCAGGCTTGGTCAAGCGCGATACAAACCATGTAGTAGGAGTCCTCCAAGTCGCCAAGCTAGGGGATCTGCTGAAAGAGGTGACAGACAAGGTAAGCAATCAGAGCTCTAAGCAATCAGTCC iieiiiii`eiiiiiiiiiiiiiiieiiiiiiiieiiiiiiiiiiiiiiiiiiiiieiiiiiiiiiiiiieiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiieiiiiiiiii`iiieeieieeieee`` AS:i:-6 XN:i:0 XM:i:1 XO:i:0 XG:i:0 NM:i:1 MD:Z:136C13 YT:Z:UU NH:i:8 CC:Z:Chr10 CP:i:18604313 HI:i:2 RG:Z:J36CK1
E00514:173:H3C3JCCXY:4:1124:12398:67234 369 Chr00 32904 0 150M Chr16 29515410 0 TCAATTTCACTTGAAGCTTACTTGTAGTTTCAGGCTTGGTCAAGCGCGATACAAACCATGTAGTAGGAGTCCTCCAAGTCGCCAAGCTAGGGGATCTGCTGAAAGAGGTGACAGACAAGGTAAGCAATCAGAGCTCTAAGCAATCAGTCC iieiiiii`eiiiiiiiiiiiiiiieiiiiiiiieiiiiiiiiiiiiiiiiiiiiieiiiiiiiiiiiiieiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiieiiiiiiiii`iiieeieieeieee`` AS:i:-6 XN:i:0 XM:i:1 XO:i:0 XG:i:0 NM:i:1 MD:Z:136C13 YT:Z:UU NH:i:8 CC:Z:Chr10 CP:i:18604313 HI:i:4 RG:Z:J36CK1
E00514:173:H3C3JCCXY:4:1124:12398:67234 369 Chr00 32904 0 150M Chr17 31040767 0 TCAATTTCACTTGAAGCTTACTTGTAGTTTCAGGCTTGGTCAAGCGCGATACAAACCATGTAGTAGGAGTCCTCCAAGTCGCCAAGCTAGGGGATCTGCTGAAAGAGGTGACAGACAAGGTAAGCAATCAGAGCTCTAAGCAATCAGTCC iieiiiii`eiiiiiiiiiiiiiiieiiiiiiiieiiiiiiiiiiiiiiiiiiiiieiiiiiiiiiiiiieiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiieiiiiiiiii`iiieeieieeieee`` AS:i:-6 XN:i:0 XM:i:1 XO:i:0 XG:i:0 NM:i:1 MD:Z:136C13 YT:Z:UU NH:i:8 CC:Z:Chr10 CP:i:18604313 HI:i:6 RG:Z:J36CK1
E00514:173:H3C3JCCXY:4:1212:19025:24532 409 Chr00 33538 0 150M * 0 0 GATTCCAAGTGCTGACTGATTGCTCTCTTTCTCCTTGTCTTGCAGGTAAGAACAAGGCCAAAGGAAAAGACAGGGAAAAAACATGAAATGAGATACTCTTGCTTTTAACCCTGATGATATGAGATATTCTTGCTCTAGTATAGCTTGTTT ii`e`ei[iiiiiiiiiiiiie[ieeieieiiiiieiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiieee`` AS:i:-12 XN:i:0 XM:i:2 XO:i:0 XG:i:0 NM:i:2 MD:Z:52T33T63 YT:Z:UU NH:i:20 CC:Z:Chr01 CP:i:11331871 HI:i:0 RG:Z:J36CK1
字段之间也就是列之间由Tab隔开,每一字段具体含义参考下图:

其中:
1. QNAME 表示reads名称;
2. FLAG:表示比对的结果,由数字表示,不同的数值含义不同,其列表如下:
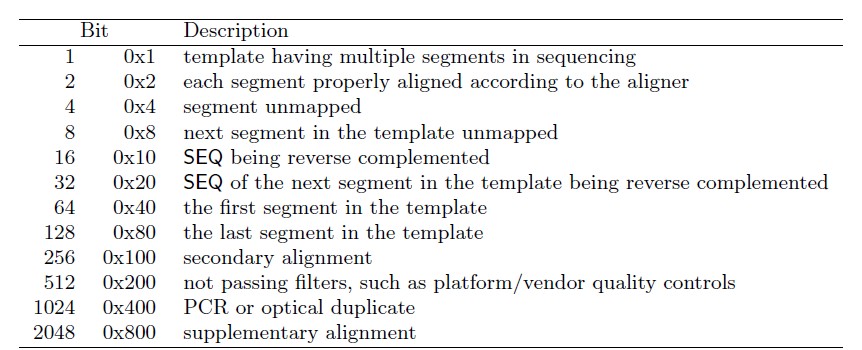
中文解释:
1 : 代表这个序列采用的是PE双端测序
2: 代表这个序列和参考序列完全匹配,没有错配和插入缺失
4: 代表这个序列没有mapping到参考序列上
8: 代表这个序列的另一端序列没有比对到参考序列上,比如这条序列是R1,它对应的R2端序列没有比对到参考序列上
16:代表这个序列比对到参考序列的负链上
32 :代表这个序列对应的另一端序列比对到参考序列的负链上
64 : 代表这个序列是R1端序列, read1;
128 : 代表这个序列是R2端序列,read2;
256: 代表这个序列不是主要的比对,一条序列可能比对到参考序列的多个位置,只有一个是首要的比对位置,其他都是次要的
512: 代表这个序列在QC时失败了,被过滤不掉了(# 这个标签不常用)
1024: 代表这个序列是PCR重复序列(#这个标签不常用)
2048: 代表这个序列是补充的比对(#这个标签具体什么意思,没搞清楚,但是不常用
比对结果数值也可以是上述数值的组合(即数值相加),如FLAG为83(64+16+2+1)表示paired-end reads中的第一个reads比对到参考序列上了;
3. RNAME:表示参考序列的名称,如基因组的染色体编号等,如果没有比对上则显示为*;
4. POS:表示比对的起始位置,以1开始计数,如果没有比对上则显示为0;
5. MAPQ:比对质量;(数字越大,特异性越高)
6. CIGAR:字符串,即比对的详细情况, 记录插入,缺失,错配,后剪切拼接的接头;
7. RNEXT:双末端测序中下一个reads比对的参考系列的名称,如果没有则用 " * " 表示,如果和前一个reads比对到同一个参考序列则用" = "表示;
8. PNEXT:下一个reads比对到参考序列上的位置,如果没有则用0表示;
9. TLEN:序列模板的长度;
10. SEQ:reads的序列信息;
11. QUAL:reads的序列质量信息;
12. 可选字段:格式如:TAG:TYPE:VALUE,其中TAG有两个大写字母组成,每个TAG代表一类信息,每一行一个TAG只能出现一次,TYPE表示TAG对应值的类型,可以是字符串、整数、字节、数组等。
常用bam/sam文件处理
由于sam格式的文件通常都非常大,所以为了节省存储空间而将sam转换为二进制格式以便于存储,也就是bam文件。 sam/bam文件可以由特定的一些软件(比如samtools)来处理的,包括格式互转、排序、建立索引等操作。
1. bam文件读取
bam文件为二进制的文件,不能直接查看,可用samtools读取:
samtools view xxx.bam
samtools view xxx.bam |less -S
2. sam/bam转换
samtools view -h xxx.bam > xxx.sam
samtools view -b -S xxx.sam > xxx.bam
3. 对bam文件排序
samtools sort xxx.bam outputPrefix
4. bam文件创建index
samtools index xxx.bam
5. 对mapping结果进行评估
在mapping之后,可以通过samtools对mapping的结果的质量进行评估。
samtools idxstats xxx.bam
执行这一步前需要经过sort和index,结果如下:
chr1 195471971 6112404 0
chr10 130694993 3933316 0
chr11 122082543 6550325 0
chr12 120129022 3876527 0
chr13 120421639 5511799 0
chr14 124902244 3949332 0
chr15 104043685 3872649 0
其中第一列是染色体名称,第二列是序列长度,第三列是mapped reads数,第四列是unmapped reads数。
6. 统计flag信息
统计bam文件中的比对flag信息,并输出比对统计结果。
samtools flagstat xxx.bam
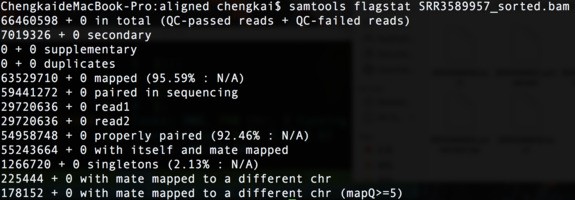
total:分析的总reads数(bam文件所有行数)
mapped:比对上的reads数(总体比对率)
paired in sequencing:成对的reads总数
read1:属于reads1的reads数量
read2:属于reads2的reads数量
properly paired:正确配对的reads数量
with itself and mate mapped:一对reads均比对上的reads数
singletons:只有单条reads比对上的reads数
以上计数均以reads条数计,一对reads计为两条。
还可以通过以下命令快速查看flag值所对应的含义:
$samtools flags 141
0x8d 141 PAIRED,UNMAP,MUNMAP,READ2
#flags值为141
#PAIRED表示这条序列采用双端测序, 其值为1;
#UNMAP表示这个序列没有mapping到参考序列上, 其值为4;
#MUNMAP表示这个序列的另一端序列没有比对到参考序列上, 其值为8;
#READ1表示这条序列是R1端序列,其值为128.
#以上数值相加和为141
7. 合并BAM文件
将多个排序后的序列文件合并为一个文件
samtools merge -n out.bam in1.bam in2.bam in3.bam…
好啦,sam/bam文件的介绍就先到这里,希望能对大家有帮助!
- 发表于 2018-06-29 09:33
- 阅读 ( 12619 )
- 分类:软件工具

