在Windows系统中安装并使用seqkit工具
seqkit软件介绍:
Seqkit是一款专门处理fsata/q序列文件的软件,由go语言编写,功能比较完善,软件使用也很稳定。
下载链接:https://bioinf.shenwei.me/seqkit/download/
下载方式:
1. 点...
seqkit软件介绍:
Seqkit是一款专门处理fsata/q序列文件的软件,由go语言编写,功能比较完善,软件使用也很稳定。
下载链接:https://bioinf.shenwei.me/seqkit/download/
下载方式:
1. 点击下载到本地(任意地址)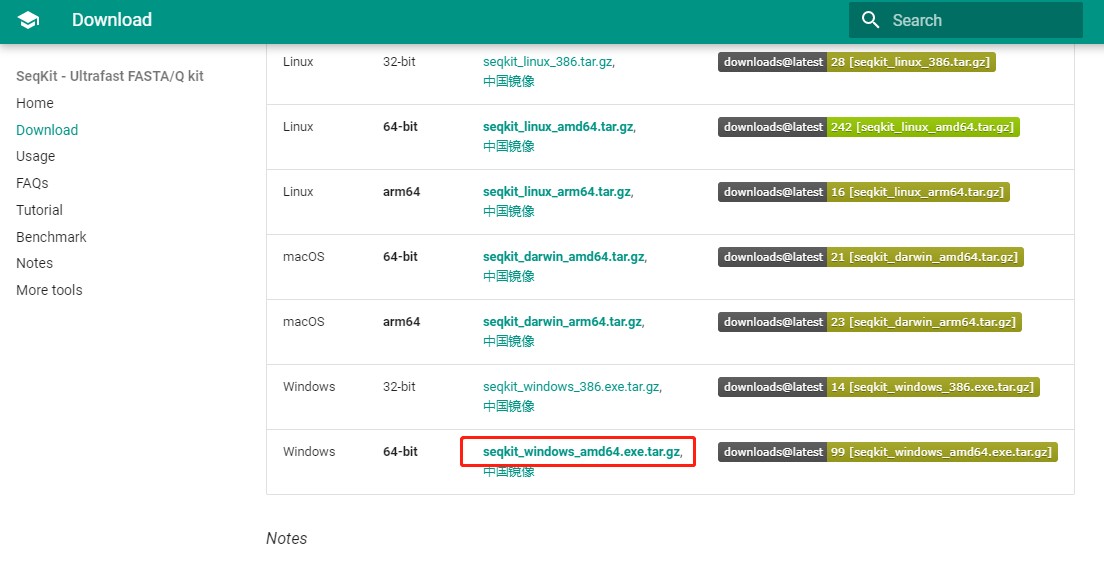
2. For windows, just copy seqkit.exe to C:\WINDOWS\system32.将下载的seqkit.exe软件复制到本地C:\Windows\System32中
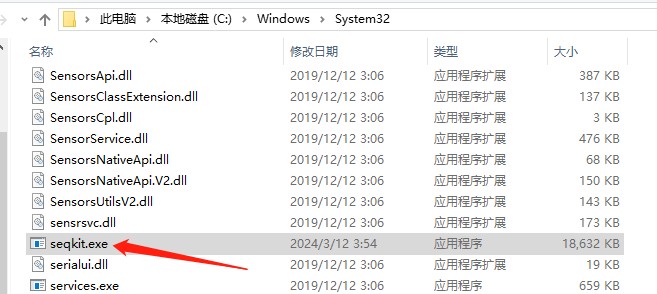 运行软件:
运行软件:
1. 打开windows系统的Windows power shell页面。输入命令:seqkit version。确认是否下载安装成功:
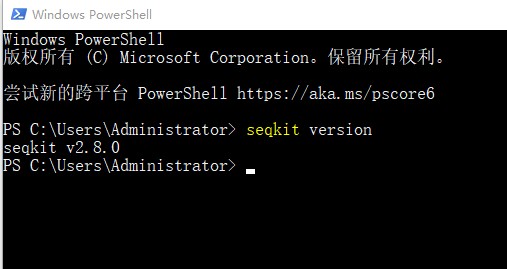 2. 之后就可以使用seqkit工具处理生信文件了。
2. 之后就可以使用seqkit工具处理生信文件了。
seqkit工具的命令如下:
Compression level:
format range default comment
gzip 1-9 5 https://github.com/klauspost/pgzip sets 5 as the default value.
xz NA NA https://github.com/ulikunitz/xz does not support.
zstd 1-4 2 roughly equals to zstd 1, 3, 7, 11, respectively.
bzip 1-9 6 https://github.com/dsnet/compress
Usage:
seqkit [command]
Commands for Basic Operation:
faidx create the FASTA index file and extract subsequences
scat real time recursive concatenation and streaming of fastx files
seq transform sequences (extract ID, filter by length, remove gaps, reverse complement...)
sliding extract subsequences in sliding windows
stats simple statistics of FASTA/Q files
subseq get subsequences by region/gtf/bed, including flanking sequences
translate translate DNA/RNA to protein sequence (supporting ambiguous bases)
watch monitoring and online histograms of sequence features
Commands for Format Conversion:
convert convert FASTQ quality encoding between Sanger, Solexa and Illumina
fa2fq retrieve corresponding FASTQ records by a FASTA file
fq2fa convert FASTQ to FASTA
fx2tab convert FASTA/Q to tabular format (and length, GC content, average quality...)
tab2fx convert tabular format to FASTA/Q format
Commands for Searching:
amplicon extract amplicon (or specific region around it) via primer(s)
fish look for short sequences in larger sequences using local alignment
grep search sequences by ID/name/sequence/sequence motifs, mismatch allowed
locate locate subsequences/motifs, mismatch allowed
Commands for Set Operation:
common find common/shared sequences of multiple files by id/name/sequence
duplicate duplicate sequences N times
head print first N FASTA/Q records
head-genome print sequences of the first genome with common prefixes in name
pair match up paired-end reads from two fastq files
range print FASTA/Q records in a range (start:end)
rmdup remove duplicated sequences by ID/name/sequence
sample sample sequences by number or proportion
split split sequences into files by id/seq region/size/parts (mainly for FASTA)
split2 split sequences into files by size/parts (FASTA, PE/SE FASTQ)
Commands for Edit:
concat concatenate sequences with the same ID from multiple files
mutate edit sequence (point mutation, insertion, deletion)
rename rename duplicated IDs
replace replace name/sequence by regular expression
restart reset start position for circular genome
sana sanitize broken single line FASTQ files
Commands for Ordering:
shuffle shuffle sequences
sort sort sequences by id/name/sequence/length
Commands for BAM Processing:
bam monitoring and online histograms of BAM record features
Commands for Miscellaneous:
merge-slides merge sliding windows generated from seqkit sliding
sum compute message digest for all sequences in FASTA/Q files
Additional Commands:
genautocomplete generate shell autocompletion script (bash|zsh|fish|powershell)
version print version information and check for update
Flags:
--alphabet-guess-seq-length int length of sequence prefix of the first FASTA record based on
which seqkit guesses the sequence type (0 for whole seq)
(default 10000)
--compress-level int compression level for gzip, zstd, xz and bzip2. type "seqkit -h"
for the range and default value for each format (default -1)
-h, --help help for seqkit
--id-ncbi FASTA head is NCBI-style, e.g. >gi|110645304|ref|NC_002516.2|
Pseud...
--id-regexp string regular expression for parsing ID (default "^(\\S+)\\s?")
-X, --infile-list string file of input files list (one file per line), if given, they are
appended to files from cli arguments
-w, --line-width int line width when outputting FASTA format (0 for no wrap) (default 60)
-o, --out-file string out file ("-" for stdout, suffix .gz for gzipped out) (default "-")
--quiet be quiet and do not show extra information
-t, --seq-type string sequence type (dna|rna|protein|unlimit|auto) (for auto, it
automatically detect by the first sequence) (default "auto")
-j, --threads int number of CPUs. can also set with environment variable
SEQKIT_THREADS) (default 4)
Use "seqkit [command] --help" for more information about a command.
- 发表于 2024-03-19 17:01
- 阅读 ( 2510 )
- 分类:软件工具
