绘制全基因组甲基化图谱
绘制全基因组甲基化图谱
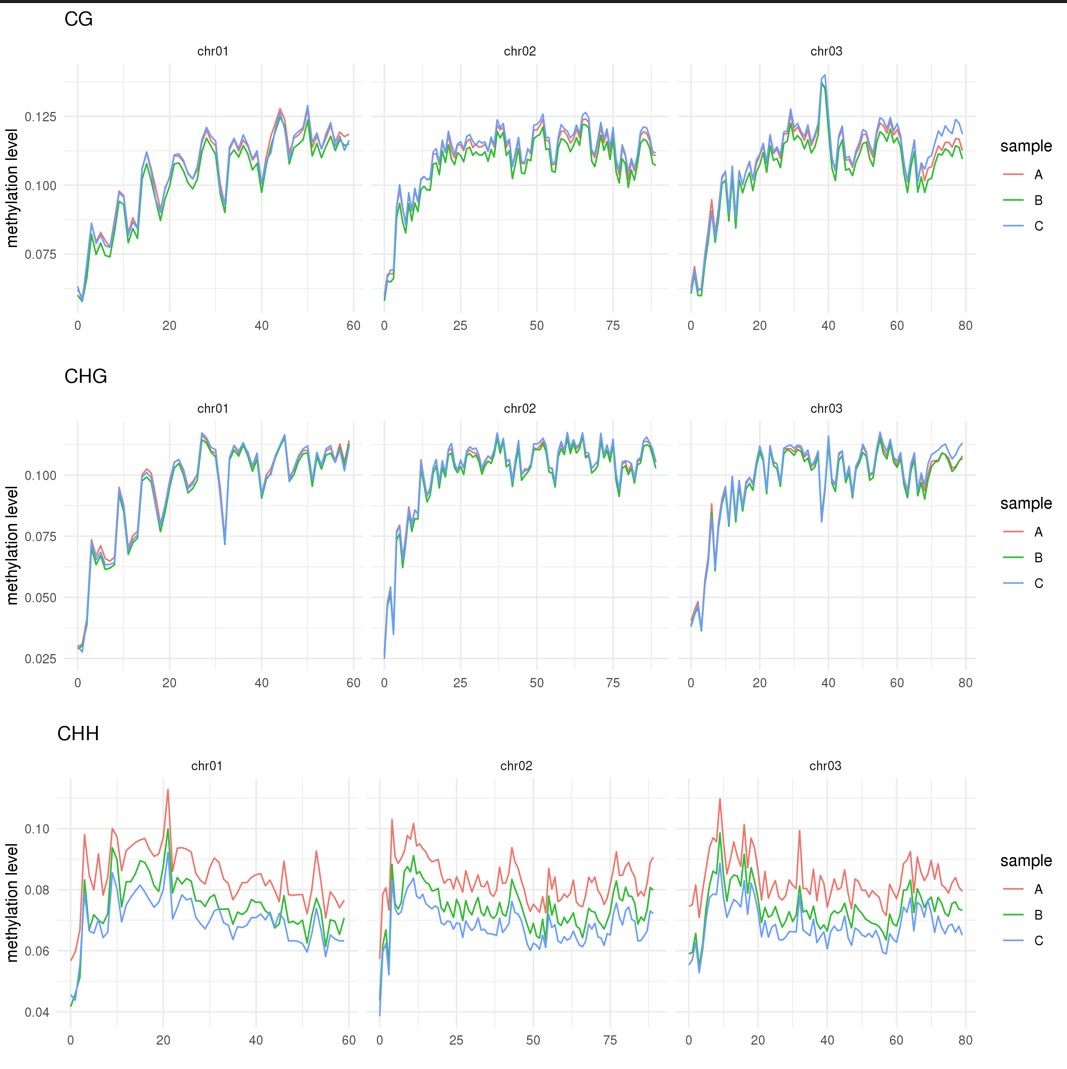
甲基化图谱可以直观的展示整个基因组甲基化的情况,绘图前我们需要准备如下文件。
#CHR start end CG CHG CHH sample
chr01 0 1000000 0.0620822859927886 0.029991922127504 0.0567165930331734 A
chr01 1000000 2000000 0.0592625465859257 0.0307502898944927 0.0595774936891281 A
chr01 2000000 3000000 0.0696789832473424 0.0414001906650762 0.0668717034822725 A
chr01 3000000 4000000 0.0860423595943484 0.0735583328046692 0.0980052745579612 A
chr01 4000000 5000000 0.0795362926184891 0.0669414899674312 0.0848156330425672 A
chr01 5000000 6000000 0.0828404829153858 0.0711317093977082 0.0799640050143304 A
chr01 6000000 7000000 0.0799346035339244 0.0658429227189838 0.0917059674275294 A
chr01 7000000 8000000 0.0775647668393782 0.0648272884283247 0.0782124352331606 A
chr01 8000000 9000000 0.0872660708335618 0.0663880113542901 0.0840268082719293 A
以1MB为窗口划分整个基因组,分别计算每个窗口内不太类型的甲基化水平。
第一列为染色体,第二列为窗口起始位置,第三列为窗口终止位置,后面分别是CG、CHG、CHH三种类型的甲基化水平,最后一列为样品或者处理。
整理好上述文件即可绘图,命令如下:
Rscript methylcytosine_level.R -i input.txt -n output -W 10 -H 10
最后附上脚本:
############################################################################
#北京组学生物科技有限公司
#author huangls
#date 2020.07.29
#version 1.0
#学习R课程:
#R 语言入门与基础绘图:
# https://zzw.h5.xeknow.com/s/2G8tHr
#R 语言绘图ggplot2等等:
# https://zzw.h5.xeknow.com/s/26edpc
###############################################################################################
library("argparse")
parser <- ArgumentParser(description='snp index manhattan plot')
parser$add_argument( "-i", "--input", type="character",required=T,
help="input smoothed data result file[required]",
metavar="filepath")
parser$add_argument( "-p", "--pallete", type="character",nargs='+',required=F,default=c("#eb65a0", "#22c2e4", "#4abb6b","#f28d21"),
help="set color pallete of plot [default=c(\"#eb65a0\", \"#22c2e4\", \"#4abb6b\",\"#f28d21\")]",
metavar="pallete")
parser$add_argument("-P", "--point.size", type="double",required=F,default=0.2,
help="the point size [optional,default:0.2]",
metavar="size")
parser$add_argument("-S", "--line.size", type="double",required=F,default=0.5,
help="the line width [optional,default:0.5]",
metavar="size")
parser$add_argument("-l", "--lab.size", type="double",required=F,default=8,
help="the font size of x and y label [optional, default:8]",
metavar="size")
parser$add_argument("-t", "--title.size", type="double",required=F,default=12,
help="the point size [optional, default: 12]",
metavar="size")
parser$add_argument("-a", "--axis.size", type="double",required=F,default=7,
help="the font size of text for axis [optional, default:7]",
metavar="size")
parser$add_argument("-T", "--title", type="character",required=F, default="",
help="the main title",
metavar="title")
parser$add_argument("-X", "--xlab", type="character", required=F,default="Chromosome",
help="the x axis lab [default Chromosome]",
metavar="label")
parser$add_argument("-Y", "--ylab", type="character", required=F,default= "ΔSNP-index",
help="the y axis lab [default ΔSNP-index]",
metavar="label")
parser$add_argument("-x", "--ylab1", type="character", required=F,default= "Bulk1 SNP-index",
help="the y axis lab of Bulk1 SNP-index [default Bulk1 SNP-index]",
metavar="label")
parser$add_argument("-y", "--ylab2", type="character", required=F,default= "Bulk2 SNP-index",
help="the y axis lab of Bulk2 SNP-index [default Bulk2 SNP-index]",
metavar="label")
parser$add_argument( "-H", "--height", type="double", default=5,
help="the height of pic inches [default 5]",
metavar="number")
parser$add_argument("-W", "--width", type="double", default=10,
help="the width of pic inches [default 10]",
metavar="number")
parser$add_argument( "-o", "--outdir", type="character", default=getwd(),
help="output file directory [default cwd]",
metavar="path")
parser$add_argument("-n", "--prefix", type="character", default="demo",
help="out file name prefix [default demo]",
metavar="prefix")
opt <- parser$parse_args()
if( !file.exists(opt$outdir) ){
if( !dir.create(opt$outdir, showWarnings = FALSE, recursive = TRUE) ){
stop(paste("dir.create failed: outdir=",opt$outdir,sep=""))
}
}
require(ggplot2)
col=c("#4197d8", "#f8c120", "#413496", "#495226", "#d60b6f", "#e66519", "#d581b7", "#83d3ad", "#7c162c", "#26755d")
#import data
data<-read.table(opt$input,header = F,comment.char = "#",fill=T)
if(ncol(data)==7){
colnames(data)=c("chr","start","end","CG","CHG","CHH","sample")
}else{
stop(paste0("data format error :",opt$input))
}
data$start = data$start/1000000;
head(data)
p1<-ggplot(data, aes(x = start, y = CG, color = sample)) +
geom_line() +
labs(title = "CG", x = "", y = "methylation level") +
theme_minimal() +
facet_wrap(~ chr, scales = "free_x", ncol = 30,shrink = TRUE)
p2<-ggplot(data, aes(x = start, y = CHG, color = sample)) +
geom_line() +
labs(title = "CHG", x = "", y = "methylation level") +
theme_minimal() +
facet_wrap(~ chr, scales = "free_x", ncol = 30,shrink = TRUE)
p3<-ggplot(data, aes(x = start, y = CHH, color = sample)) +
geom_line() +
labs(title = "CHH", x = "", y = "methylation level") +
theme_minimal() +
facet_wrap(~ chr, scales = "free_x", ncol = 30, shrink = TRUE)
library(gridExtra)
png(filename=paste0(opt$outdir,"/",opt$prefix,".png"), height=opt$height*300, width=opt$width*300, res=300, units="px")
grid.arrange(p1, p2, p3, nrow = 3)
dev.off()
pdf(file=paste0(opt$outdir,"/",opt$prefix,".pdf"), height=opt$height, width=opt$width)
grid.arrange(p1, p2, p3, nrow = 3)
dev.off()
- 发表于 2024-07-25 14:08
- 阅读 ( 1073 )
- 分类:R
