组装"预实验" | 基因组调研图
基因组调研图有助于了解即将组装的这个物种的基因组复杂程度,为后续的正式的基因组组装去提供一些依据。基因组的复杂程度可以从基因组大小,倍性,杂合度,重复率等方面去进行评价。
用什么来...
基因组调研图有助于了解即将组装的这个物种的基因组复杂程度,为后续的正式的基因组组装去提供一些依据。基因组的复杂程度可以从基因组大小,倍性,杂合度,重复率等方面去进行评价。
用什么来做基因组调研图?
基因组的调研图一般都会使用kmer分析来实现。kmer分析通常都是基于30到80×的二代测序数据去完成。
如何预估自己所需测序数据量?
1. 流式细胞仪:
通过测量细胞核的DNA含量来估算基因组大小,1pg=978M;
2. 网站查询:
植物基因组大小查询:https://cvalues.science.kew.org/
动物基因组大小查询:http://www.genomesize.com/
什么是k-mer?
k-mer是指包含在一段序列中的长度为k的子串。
现有一段序列,长度是15个碱基,把k的长度设置成5,这意味着需要从这段序列上,每隔一个碱基取一个长度为5个碱基的序列片段。那么序列长度为15的序列可以取11个5mer。如图:
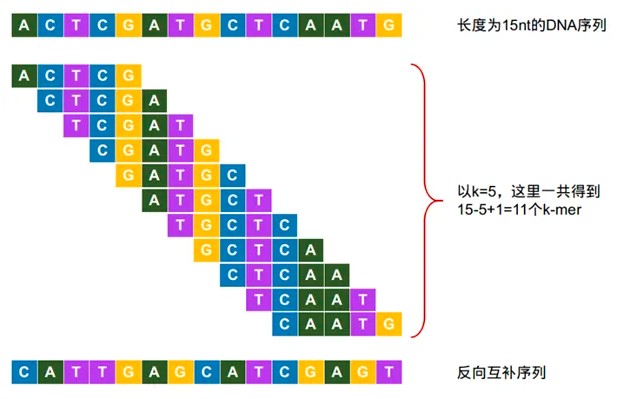
针对长度为K的K-mer,对于 A,T,C,G四种碱基类型,一共能产生的K-mer种类数为4^k个。
如何利用k-mer评估基因组大小?
1.获取k-mer频数分布表(假设样本名为A)
ls /Data/A* | awk '{ print "gzip -dc " $0}' > A_generate.file
jellyfish count -t 20 -C -m 21 -s 3G -g A_generate.file -G 2 -o A.sif
# jellyfish count:k-mer计数
# -t 指定线程数
# -C 对DNA正负链都进行统计,表示考虑DNA正义与反义链。如果是双端测序reads,需要这个参数。
# -m k-mer长度设置为21bp.
# -s 3G 存储用的hash表大小为3G,这个参数识别单位M(Mbp)和G(Gbp)。如果设置不够大,会生成多个hash文件。最好设置的值大于总的独特的(distinct)k-mer数。如果基因组大小为G,每个reads有一个错误,总共有n条reads,则该值可以设置为[(G + n)/0.8]。
# -g --generator=path 记录产生fast[aq]命令的文件
# -G 同时运行的数目
# -o 指定输出文件的名字,hash格式储存的k-mer频数文件
2.统计k-mer频率
jellyfish histo -v -t 20 -h 10000000 -o A.histo A.sif
# 结果最后是两列,第一列是x,表示的是出现的次数;第二列是y,表示出现x次数的kmer数目。
# -v 显示详细信息
# -t 线程数
# -h x的最大值,默认是10000
# -o 输出文件的名字。
3. 统计k-mer
jellyfish stats A.sif -o A.stats
结果有四行:
Unique: 只出现过一次的k-mer数量
Distinct: 特异的k-mer数目
Total: k-mer总数
Max_count: 频数最高的k-mer数量
4. 基因组特征评估
genomescope.R -i A.histo -o genomescope_A -p 2 -k 21 -m 10000000
# -i 输入文件
# -o 输出文件夹
# -p 倍性
# -k k-mer长度
# -m 最大k-mer覆盖度,覆盖度大于该数字的k-mer将被忽略
结果解读
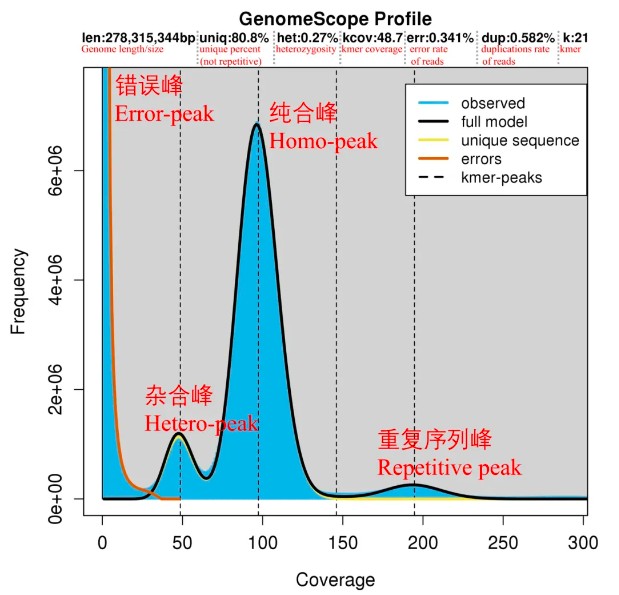
在输出文件夹中会存在四个图片,三个txt文件。其中最重要的结果图片是 transformed_linear_plot.png,图中信号含义标注如下图,其中kcov指的是杂合峰的覆盖度:
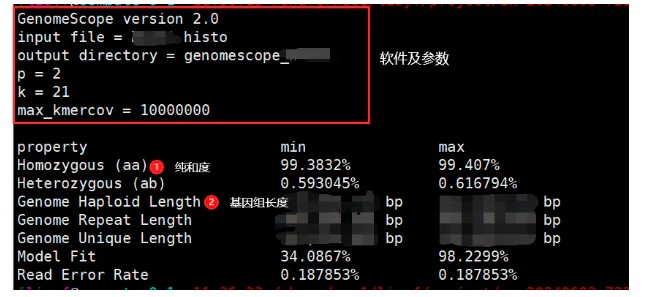
最重要的结果文件是 summary.txt,其中信息如下图
- 发表于 2024-07-30 15:39
- 阅读 ( 1887 )
- 分类:基因组学
