TWAS分析的原理和工具
TWAS的分析原理
人类复杂形状的出现很大程度上可以归因于遗传变异,但遗传变异对于性状的作用机制却尚不明确。许多遗传变异通过调节基因表达来影响复杂性状,即改变一种或多种蛋白质的丰度。有人认为这种关系可以通过关联扫描来进行研究,然而,测量基因表达的研究受到样本可用性和成本的阻碍,少数已发表的表达和复杂性状研究比单独研究性状要小几个数量级。因此,许多表达-性状关联无法检测到,尤其是那些影响较小的关联。为了减轻样本量小导致的功效降低,最好的解决办法是选取影响基因表达的遗传变异(表达数量性状位点,eQTL)与在GWAS分析中确定的性状相关变异的交集。然而,这种方法也有一定的弊端,同样可能错过较弱的表达-性状关联。
TWAS(Transcriptome-wide Association Study)是一种用于研究基因表达和疾病之间关系的方法,通过对全基因组的基因表达数据进行统计分析,以识别与特定疾病或性状相关的基因表达变异。TWAS能够很好的解决上述分析方法中存在的“数据集之间存在巨大的样本差异的问题”,这主要是由于其分析方法的不同:
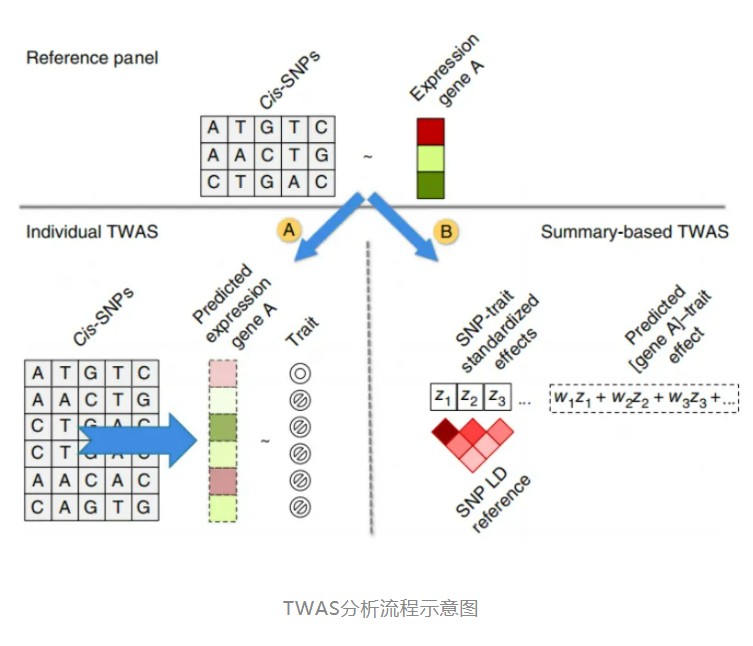
TWAS分析的第一步,基于reference panel来建模,构建SNP和基因表达量之间的关系。reference panel中的样本同时拥有基因分型和表达量的结果,根据距离确定基因对应的SNP位点,比如选择基因上下游500kb或者1Mb范围内的SNP位点,拟合这些SNP位点和基因表达量之间的关系,通常,用于构建表达量模型的数据集可以较小。
第二步,用第一步建模的结果来预测另外一个队列的基因表达量,这个队列中的样本量只有GWAS结果,称之为gwas cohort, 这一步可以看做是对gwas cohort中的基因表达量进行填充,需要注意的是,这里分析用到的变异位点应为表达量模型构建时位点的子集。
第三步,用填充之后的基因表达量来分析基因和性状之间的关联。
下图对TWAS的分析流程进行了很好的解释:
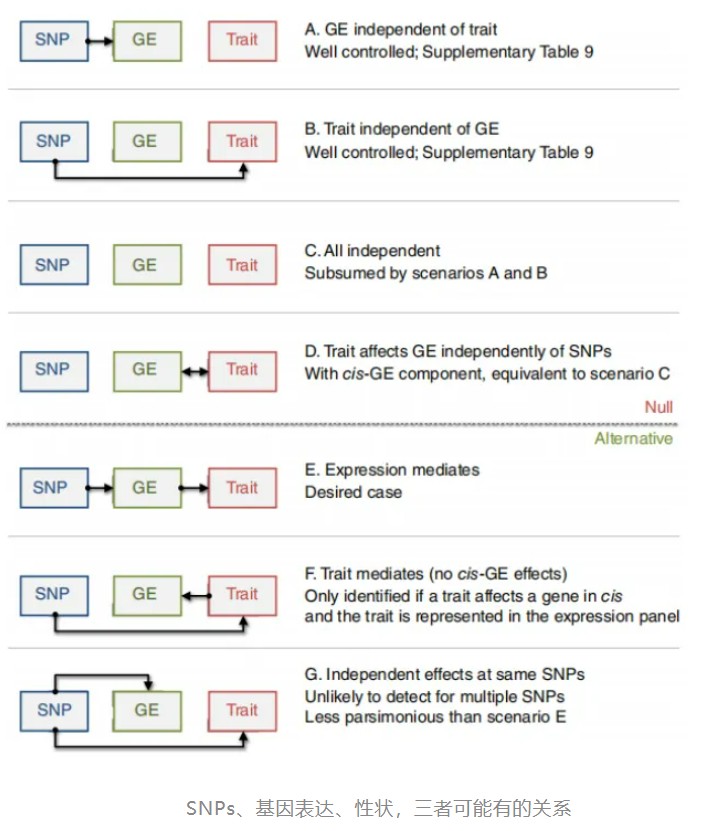
 遗传标记(SNPs;蓝色)、基因表达(GE;绿色)和性状(Trait;红色)之间可能的因果关系模式有如下几种。场景A-D将被TWAS模型视为不具有明显的关联关系。E-G可以被确定为遗传标记、基因表达和性状之间的关联是显著性的。
遗传标记(SNPs;蓝色)、基因表达(GE;绿色)和性状(Trait;红色)之间可能的因果关系模式有如下几种。场景A-D将被TWAS模型视为不具有明显的关联关系。E-G可以被确定为遗传标记、基因表达和性状之间的关联是显著性的。
 TWAS的分析软件
TWAS的分析软件
我们主要介绍两款常用的TWAS分析软件:
1.Predixcan
Gamazon,E.,Wheeler,H.,Shah,K.et al.A gene-based association method for mapping traits using reference transcriptome data. Nat Genet 47, 1091–1098 (2015).
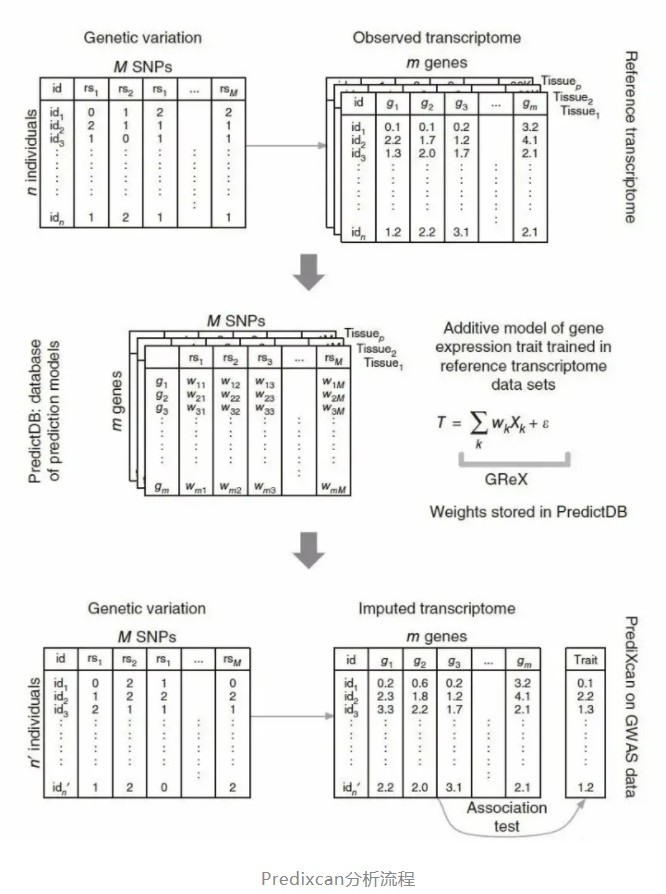
 Predixcan是于2015年由一群芝加哥学者研发的,适用于个体级别的GWAS数据的TWAS分析软件,作者认为基因表达水平受到三个因素的调控,其中主要的两个是遗传因素和疾病状态,PrediXcan的目的是建立起受遗传调控的基因表达与性状之间的关系。整个工作流程和主流的TWAS分析流程一致,主要分为两步:(1)估算SNP调控的基因表达水平:借助类似于机器学习的思想,利用GTEx Project,GEUVADIS和DGN数据库中基因型数据和基因表达数据做训练集,然后估算用户导入的基因型数据中缺失的表达数据。一旦得到表达数据,就可建立起基因表达与性状之间的关系。(2)建立基因表达水平与性状之间的关联。
Predixcan是于2015年由一群芝加哥学者研发的,适用于个体级别的GWAS数据的TWAS分析软件,作者认为基因表达水平受到三个因素的调控,其中主要的两个是遗传因素和疾病状态,PrediXcan的目的是建立起受遗传调控的基因表达与性状之间的关系。整个工作流程和主流的TWAS分析流程一致,主要分为两步:(1)估算SNP调控的基因表达水平:借助类似于机器学习的思想,利用GTEx Project,GEUVADIS和DGN数据库中基因型数据和基因表达数据做训练集,然后估算用户导入的基因型数据中缺失的表达数据。一旦得到表达数据,就可建立起基因表达与性状之间的关系。(2)建立基因表达水平与性状之间的关联。
2.FUSION
Gusev et al. “Integrative approaches for large-scale transcriptome-wide association studies” 2016 Nature Genetics

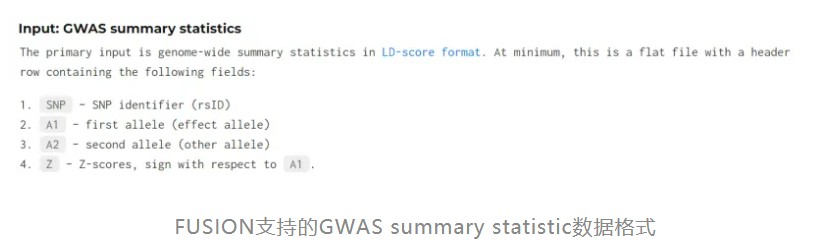
FUSION是另一款TWAS分析工具。其分析思想与Predixcan大致相似,第一步是构建功能/分子表型的遗传成分的预测模型(该软件可提供来自多项研究的预先计算的预测模型以促进分析),不同的是FUSION使用GWAS summary statistic数据与表型做关联分析。summary statistic顾名思义,是对GWAS数据的一个概括总结,包含了结果中最核心的信息,这类格式的GWAS数据通常来源于已经公开发表的文章和各类数据库中。
- 发表于 2024-11-12 17:29
- 阅读 ( 2316 )
- 分类:软件工具
