Mfuzz做时间聚类分析
Mfuzz做时间聚类分析
Mfuzz 是 R 语言中的一个用于聚类分析的包,专为时间序列或条件数据的基因表达分析而设计。它广泛应用于生物信息学和基因组学研究中,尤其适用于处理具有时间序列的数据集,比如基因表达时间序列。
Mfuzz 使用模糊 C均值聚类算法,将数据点分配到多个聚类类别中,同时计算每个数据点的成员隶属度。与传统的硬聚类方法不同,模糊聚类允许数据点属于多个聚类类别,这对于基因表达数据非常有意义,因为某些基因可能同时参与多个生物过程。
通过 Mfuzz,研究人员能够发现动态的基因表达模式,深入理解基因在不同条件或时间点的调控过程。这对于理解复杂的生物学过程(如发育、疾病进程和环境应答)非常有帮助。
1.加载需要的包
# 1.安装CRAN来源常用包
#设置镜像,
local({r <- getOption("repos")
r["CRAN"] <- "http://mirrors.tuna.tsinghua.edu.cn/CRAN/"
options(repos=r)})
# 依赖包列表:自动加载并安装
package_list <- c("ggplot2","Mfuzz","dplyr","devtools")
# 判断R包加载是否成功来决定是否安装后再加载
for(p in package_list){
if(!suppressWarnings(suppressMessages(require(p, character.only = TRUE, quietly = TRUE, warn.conflicts = FALSE)))){
install.packages(p, warn.conflicts = FALSE)
suppressWarnings(suppressMessages(library(p, character.only = TRUE, quietly = TRUE, warn.conflicts = FALSE)))
}
}
2.读入数据
#设置工作路径
setwd("~/work/10.mfuzz")
## 导入基因表达量
gene <- read.table("gene_fpkm.txt",header =T,row.names=1,sep="\t", check.names = F)
head(gene)
基因表达量数据格式如下:
ID 0h 1h 2h 3h 4h ENSGALG00000000003 0.0000000 0.01421203 0.08885999 0.01337165 0.0000000 ENSGALG00000000011 11.3414235 8.40606978 9.13251124 8.91866821 9.5756292 ENSGALG00000000038 0.1329301 0.09363415 0.00000000 0.00000000 0.0000000 ENSGALG00000000044 0.0000000 0.00000000 0.00000000 0.00000000 0.0000000 ENSGALG00000000048 NA 1.00392485 1.43193841 1.06263161 0.9645617 ENSGALG00000000055 56.8308552 74.88416519 59.40479219 98.77510726 95.3780266
3.数据处理
#参数
#目标聚类群的个数
n = 6
# 设置图形布局,2 行 5 列
mfrow = c(2, 3) # 设置图形布局,2 行 5 列
## 转换格式
gene.m <- data.matrix(gene)
gene.n <- new("ExpressionSet",exprs = gene.m)
## 过滤缺失超过25%的基因
gene.f <- filter.NA(gene.n, thres=0.25)
## 3 genes excluded.
## mean填补缺失
gene.i <- fill.NA(gene.f,mode="mean")
## 过滤标准差为0.1的基因,表达变化不大
gene.o <- filter.std(gene.i,min.std=0.1,visu = T)
## 69 genes excluded.
## 标准化
gene.s <- standardise(gene.o)
## 聚类,需手动定义目标聚类群的个数
cl <- mfuzz(gene.s,c=n,m=mestimate(gene.s))
## 输出基因ID
write.table(cl$cluster,"output.txt",quote=F,row.names=T,col.names=F,sep="\t")
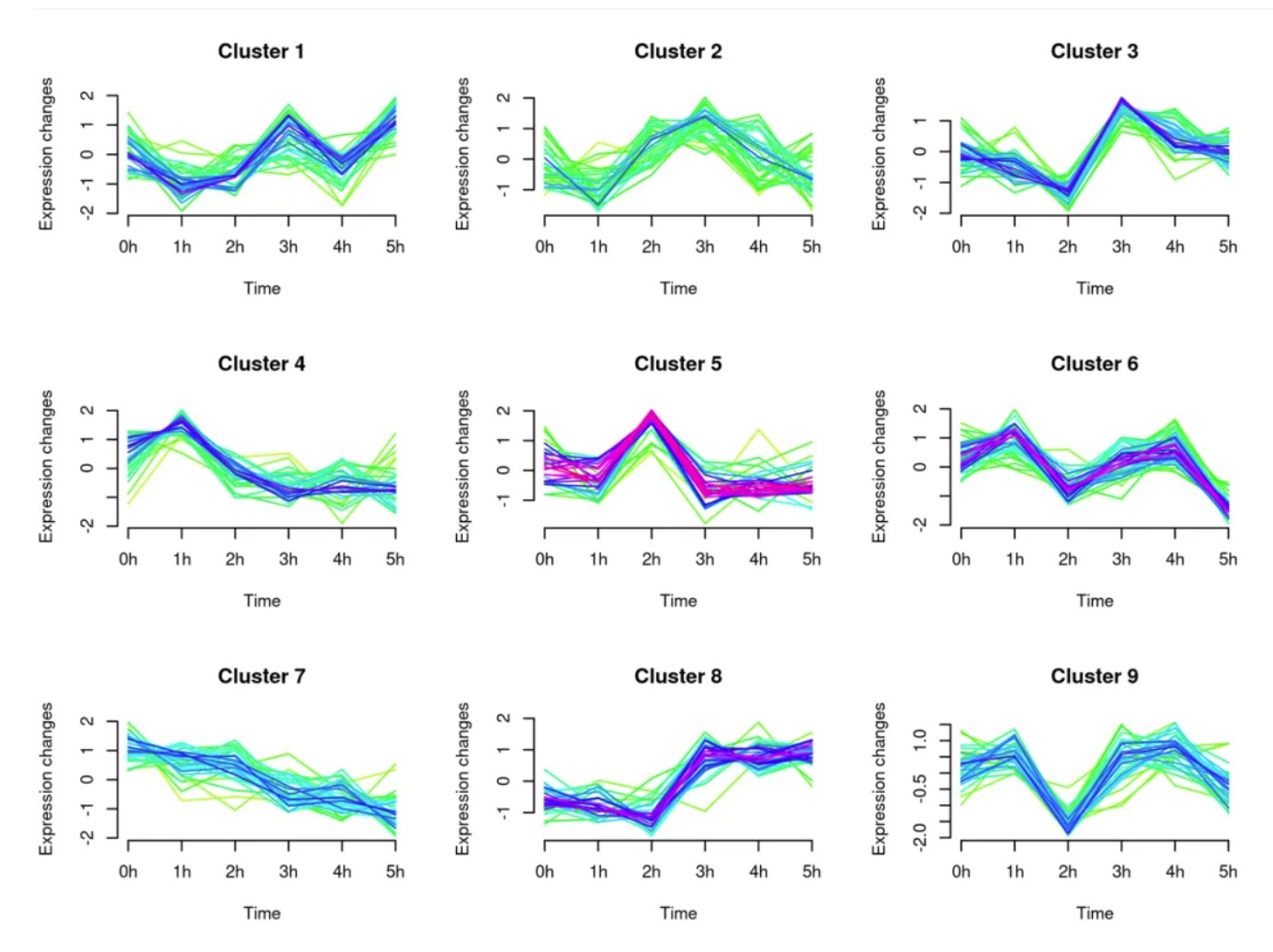
4.绘图
由于使用Mfuzz自带的绘图函数报错,改用R语言自带的plot函数进行绘图。
clusterindex <- cl[[3]] # 各基因所属cluter
memship <- cl[[4]] # 各基因所属cluter的隶属度
memship[memship < 0] <- -1 # 将小于最低隶属度0的隶属度强制改为-1
colorindex <- integer(dim(exprs(gene.s))[[1]]) # 行数长度的0向量
## 配色
fancy.blue <- c(c(255:0), rep(0, length(c(255:0))),
rep(0, length(c(255:150))))
fancy.green <- c(c(0:255), c(255:0), rep(0, length(c(255:150))))
fancy.red <- c(c(0:255), rep(255, length(c(255:0))),
c(255:150))
colo <- rgb(b = fancy.blue/255, g = fancy.green/255,
r = fancy.red/255) # 取出颜色
length(colo) # 618
## [1] 618
# 根据隶属度范围0-1,生成与颜色向量等长的数值向量
colorseq <- seq(0, 1, length = length(colo)) # 取出与颜色一致长度的值
# 基础绘图布局
par(mfrow = mfrow, bg = "white", col.axis = "black", # 设置图形布局,2 行 5 列
col.lab = "black", col.main = "black", col.sub = "black",
col = "black")
## 绘制折线图
vector <-c(1:n)
for (j in vector) {
tmp <- exprs(gene.s)[clusterindex == j, , drop = FALSE] # 时序输入文件,标准后的表达矩阵
tmpmem <- memship[clusterindex == j, j] # 取出cluster j中的membership
# 坐标轴数值
ymin <- min(tmp) # 定义最值
ymax <- max(tmp)
xlim.tmp <- c(1, dim(exprs(gene.s))[[2]]) # 列数
ylim <- c(ymin, ymax)
# 绘制坐标轴标签和标题
plot.default(x = NA, xlim = xlim.tmp, ylim = ylim, # 绘制坐标轴标签和标题
xlab = "Time", ylab = "Expression changes", main = paste("Cluster",
j), axes = FALSE )
# 绘制坐标轴
axis(1, 1:dim(exprs(gene.s))[[2]], colnames(gene.i), # 绘制坐标轴
col = "black")
axis(2, col = "black")
# 根据membership数值给基因配色,即大致相同的隶属度,在各Cluter中绘图基本是相似的。
for (jj in 1:(length(colorseq) - 1) ) {
tmpcol <- (tmpmem >= colorseq[jj] & tmpmem <=
colorseq[jj + 1]) # 通过membership给予颜色
if (sum(tmpcol) > 0) {
tmpind <- which(tmpcol)
for (k in 1:length(tmpind)) {
# if (missing(time.points)) {
lines(tmp[tmpind[k], ], col = colo[jj]) # 划线数据为tmp,标准化后的表达量值。
# }
# else lines(time.points, tmp[tmpind[k], ],
# col = colo[jj])
}
}
}
}
par(mfrow = c(1, 1))
结果图如下:
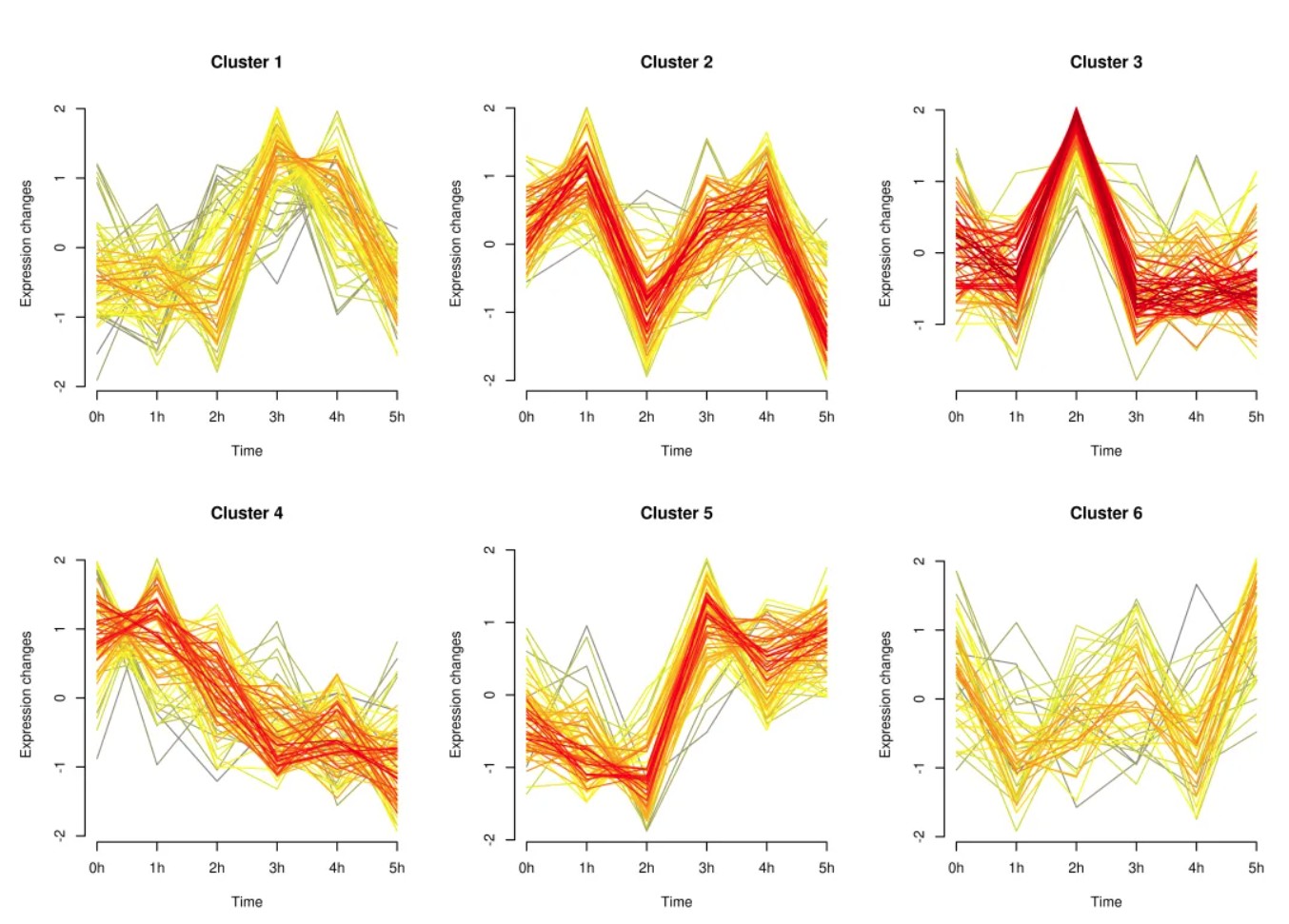
好了,小编就先给大家介绍到这里。希望对您的科研能有所帮助!祝您工作生活顺心快乐!
- 发表于 2025-01-02 16:23
- 阅读 ( 5327 )
- 分类:软件工具
