如何计算基因组中的连锁不平衡
连锁不平衡(linkage disequilibrium,LD)是指在某一个群体中,不同位置上的两个基因座同时遗传的频率明显高于预期的随机频率现象,一般用D,D'和r²来表示LD的程度。
连锁不平衡(linkage disequilibrium,LD)是指在某一个群体中,不同位置上的两个基因座同时遗传的频率明显高于预期的随机频率现象,一般用D,D'和r²来表示LD的程度。D(连锁不平衡系数)是最基础的,D衡量的是实际观察到的单倍型频率与期望频率的差异。比如说,两个位点A和B,各自的等位基因频率分别是P(A)、P(a)和P(B)、P(b)。如果两个位点独立,那么单倍体AB的期望频率应该是P(A)P(B),而实际观察到的频率是P(AB)。D就是P(AB)-P(A)P(B)。不过D的取值范围可能受等位基因频率的影响,所以可能存在不同的最大值和最小值,标准化的不平衡系数D'能够避免这种对等位基因频率的依赖。

D'是标准化的D。D'的计算方法是将D除以其理论上的最大值。D'的优点是消除了等位基因频率的影响,使得不同位点之间的比较更可行。但在某些情况下,D'可能会有偏差:比如当单倍型为2或3种时,|D'|一定等于1,但是当|D'|<1时,D'的值究竟表示多大程度的连锁不平衡,是很难做出准确判断的。另外D'严格依赖于样品的大小,如果样本偏少时,snp数量比较少,这样算出来的D'就会偏大,尤其是某个位点其中一个等位基因频率很低时,因此较高D'背后,实际上可能是连锁不平衡程度很低的两个位点。
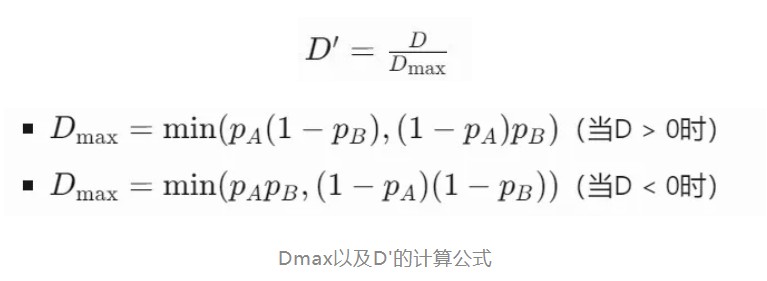 r²,也就是相关系数的平方,根据其计算公式,r²的取值范围在0到1之间,r²越大,说明两个位点的关联性越强。它的优点是不受等位基因频率的影响,但可能对样本量敏感,样本量小的时候估计可能不准确。
r²,也就是相关系数的平方,根据其计算公式,r²的取值范围在0到1之间,r²越大,说明两个位点的关联性越强。它的优点是不受等位基因频率的影响,但可能对样本量敏感,样本量小的时候估计可能不准确。 对三种系数在衡量和计算LD时的方法进行比较,我们能够清晰的看出三种参数在使用时的区别:D的原始值可能难以直接比较不同位点对,因为其范围受等位基因频率影响,而D'和r²都是标准化的,但方式不同。D'关注的是D相对于其可能的最大值,而r²则类似于统计学中的决定系数,反映两个位点的相关性。在优缺点方面,D的缺点可能是难以跨不同频率的位点比较,但能直接反映连锁不平衡的方向和强度。D'能标准化,但可能在低频等位基因时高估连锁不平衡的程度,比如当某个单倍型频率很低时,即使实际关联不强,D'也可能接近1。而r²对样本量敏感,但能更好地反映位点间的预测能力,比如在关联分析中,r²高说明一个位点能较好地代表另一个位点。在实际的数据分析中,D'可能更适合用于确定历史重组事件,因为它能反映是否存在完全的连锁不平衡,可能用于识别LD block的边界,因为当D'下降时,意味着可能有重组事件发生。而r²可能更适合衡量两个位点之间的连锁强度,尤其是在关联研究中,因为r²高意味着标记位点能有效代表目标位点,比如在GWAS中选择tag SNP时,r²更重要。
对三种系数在衡量和计算LD时的方法进行比较,我们能够清晰的看出三种参数在使用时的区别:D的原始值可能难以直接比较不同位点对,因为其范围受等位基因频率影响,而D'和r²都是标准化的,但方式不同。D'关注的是D相对于其可能的最大值,而r²则类似于统计学中的决定系数,反映两个位点的相关性。在优缺点方面,D的缺点可能是难以跨不同频率的位点比较,但能直接反映连锁不平衡的方向和强度。D'能标准化,但可能在低频等位基因时高估连锁不平衡的程度,比如当某个单倍型频率很低时,即使实际关联不强,D'也可能接近1。而r²对样本量敏感,但能更好地反映位点间的预测能力,比如在关联分析中,r²高说明一个位点能较好地代表另一个位点。在实际的数据分析中,D'可能更适合用于确定历史重组事件,因为它能反映是否存在完全的连锁不平衡,可能用于识别LD block的边界,因为当D'下降时,意味着可能有重组事件发生。而r²可能更适合衡量两个位点之间的连锁强度,尤其是在关联研究中,因为r²高意味着标记位点能有效代表目标位点,比如在GWAS中选择tag SNP时,r²更重要。
计算位点之间的LD
位点之间的LD,也就是计算r²,我们可以使用plink软件进行:#将vcf文件转换成plink格式
plink --vcf yourfile.vcf --recode --out yourfile --allow-extra-chr
#计算位点之间的r2
#--ld-window-kb 250 对距离在250kb之内的SNP位点进行分析(默认为1Mb)
#--ld-window-r2 0.2 仅输出r2高于该参数值的LD分析结果(仅适用于--r2)(缺省为0.2)
# 若要报告所有对,则将--ld-window-r2设置为0
plink --file yourfile --r2 --ld-window-kb 250 --ld-window-r2 0.1 -out yourfile --allow-extra-chr
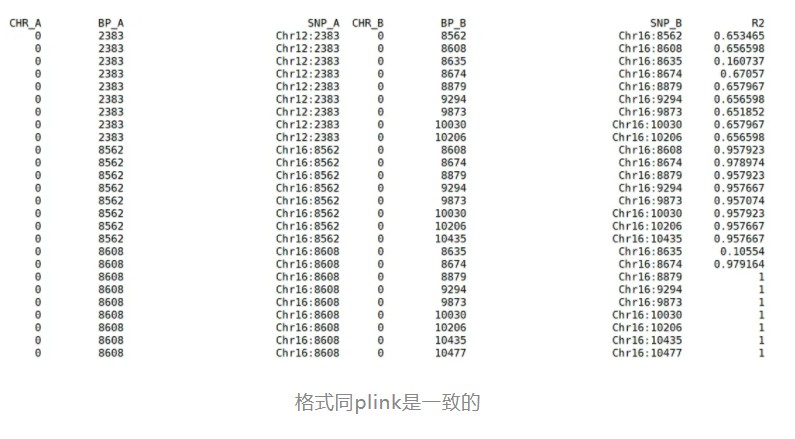
最后会输出一个ld后缀的结果文件:

除此之外,也可以使用Haploview:
#-nogui 结果以非交互的形式输出
#-maxdistance 250 对距离在250kb之内的SNP位点进行分析(默认为500kb)
#-minMAF 0.05 标记的最小次要等位基因频率为0.05(默认是.001)
#-missingCutoff 0.1 排除缺失数据比例超过10%的个体(默认是.5)
java -jar Haploview.jar -nogui -pedfile yourfile.ped -out yourfile -maxdistance 250 -minMAF 0.05 -missingCutoff 0.1 -hwcutoff 0 -dprime
同样最后会得到一个后缀为.ld的结果文件:
LD block估计
计算LD block,我们一般使用的是plink软件的block功能:
#使用plink生成bed格式的基因型文件
plink --file yourfile --make-bed --out yourfile --allow-extra-chr
#--blocks 进行LD block推断
#--blocks-max-kb 指定LD block的最大范围(默认200kb)
plink --bfile yourfile --blocks no-pheno-req --blocks-max-kb 250 -out yourfile --allow-extra-chr
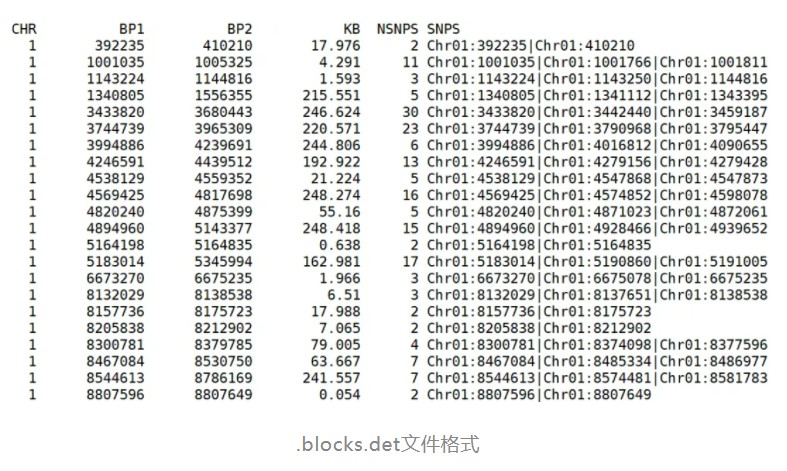
能够得到两个结果文件,首先是简化的LD blocks结果文件,每一行为一个LD block,包含了block里面的所有snp位点的id信息,该文件为.blocks结尾:

- 发表于 21小时前
- 阅读 ( 38 )
- 分类:遗传进化
