linux下samtools安装指南
序列比对(将测序reads与已知序列信息的基因或基因组进行比对)是高通量测序数据分析中最重要的一环,无论是转录组还是重测序都是基于比对结果来进行后续各项分析的,比对结果格式比较常见的是sam和bam文件,例如转录组Tophat分析软件输出的比对结果为.bam文件,而重测序中BWA、bowtie等比对软件则主要输出为.sam文件。
samtools是一个用于操作sam和bam文件的工具软件,能够对比对文件进行二进制查看、格式转换、排序及合并等,结合sam格式中的flag、tag等信息,还可以完成比对结果的统计汇总,是处理sam和bam文件不可或缺的神器!
最近小编也在学习使用这款软件,但安装时却遇到了很大麻烦;由于安装前没有仔细查看安装文档,而是下载后直接就开始安装了,安装时一直报错,才发现samtools是依赖很多包的,所以小编在这里整理了一下安装步骤,也好方便大家安装。
首先看一下samtools需要安装哪些库或是包:
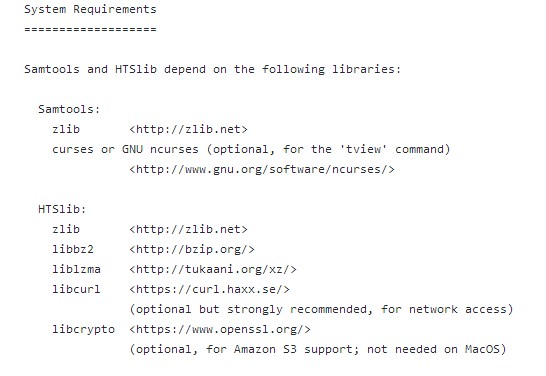
可以看到需要的包还是挺多的。没关系,我们一个一个安装即可。(小编将软件安在根目录下的biosoft目录。其中包或者库都是默认安装的,需要root权限,只有samtools是安装在/biosoft/samtools/samtools-v1.9下。)
zlib库安装
下载并解压:
mkdir /biosoft/zlib
cd /biosoft/zlib
curl -O http://www.zlib.net/zlib-1.2.11.tar.gz
tar xvfz zlib-1.2.11.tar.gz
cd zlib-1.2.11
进入解压后的zlib目录,执行以下命令安装zlib
./configure
make
make check
make install
在make install这一步,由于要把zlib安装到/usr/local/lib 路径下,所以可能需要root 权限。安装成功后,可以在/usr/local/lib下找到 libz.a。zlib安装较为简单,通常可以顺利的安装成功。
curses库安装
接下来要安装curses库,这里可以使用命令直接安装。
首先查看curses相关的安装包:
yum search curses
搜索结果如下图所示:
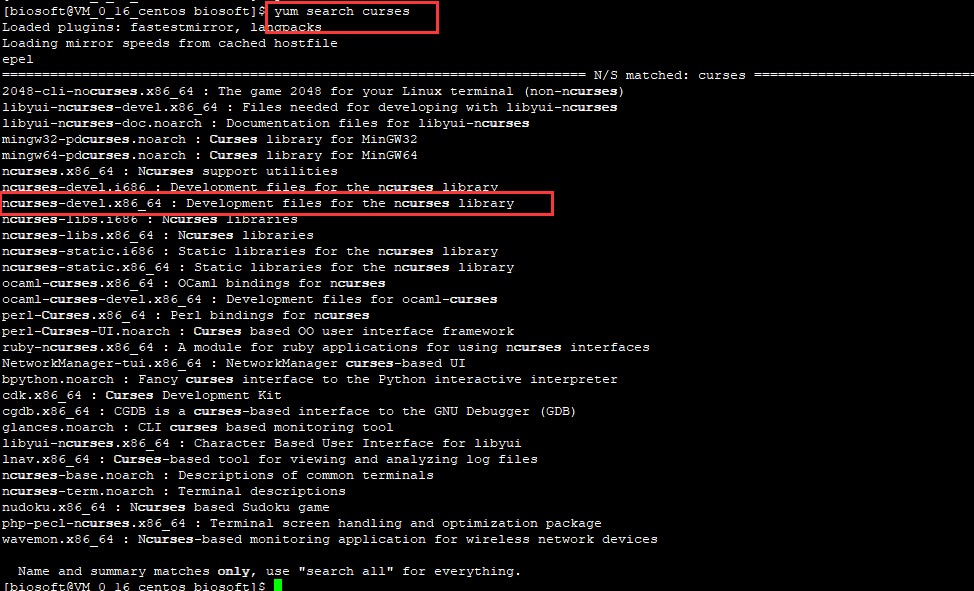
找到这个安装文件ncurses-devel.x86_64,安装即可:
yum -y install ncurses-devel.x86_64
htslib包安装
再之后需要安装htslib包。
下载:
mkdir /biosoft/htslib
cd /biosoft/htslib
wget https://github.com/samtools/htslib/archive/develop.zip
解压:
unzip develop.zip
cd htslib-develop
安装指令:
autoconf #Generate the configure script, if needed
./configure #Optional, needed for choosing optional functionality
make
make install
在./configure过程中可能会报错,原因是有些库或包没安装,根据报错信息提示的将缺失包安装即可。全部使用yum安装,以bzip2为例,系统提示缺失该库,yum search bzip2搜索相关安装包,如下:
yum search bzip2
在搜索结果中选择devel.x86_64结尾的文件进行安装,该安装包包含库的所有文件,如头文件等,而其余安装包只包含部分文件。
yum –y install bzip2-devel.x86_64
安装完成后,再./configure一次,如果提示缺少库或包,按照前面方法安装即可,直到全部安装成功,然后进行编译。
make
make install
samtools安装
最后就可以安装samtools了,samtools官网地址:https://github.com/samtools/samtools
下载并解压:
mkdir /biosoft/samtools
cd /biosoft/samtools
wget https://github.com/samtools/samtools/archive/develop.zip
unzip develop.zip
mkdir samtools-v1.9
cd samtools-develop
编译安装:
autoheader # Build config.h.in (this may generate a warning about # AC_CONFIG_SUBDIRS - please ignore it).
autoconf -Wno-syntax # Generate the configure script
./configure --prefix=/biosoft/samtools/samtools-v1.9 # --prefix 添加安装路径
make
make install
由于之前的准备工作都做完了,所以这时候安装samtools就会很顺利了,小编也没有再遇到任何的报错信息。到这里samtools的安装就完成了。希望小编的努力对大家有所帮助!
最后祝您科研愉快!
- 发表于 2018-11-01 10:03
- 阅读 ( 9324 )
- 分类:软件工具
