NCBI基因组数据格式修改
NCBI基因组数据格式修改
绝大数生信分析都要用到参考基因组数据,但有些基因组的数据会让人十分头疼,例如NCBI上的基因组数据。

其gff文件是这样的:
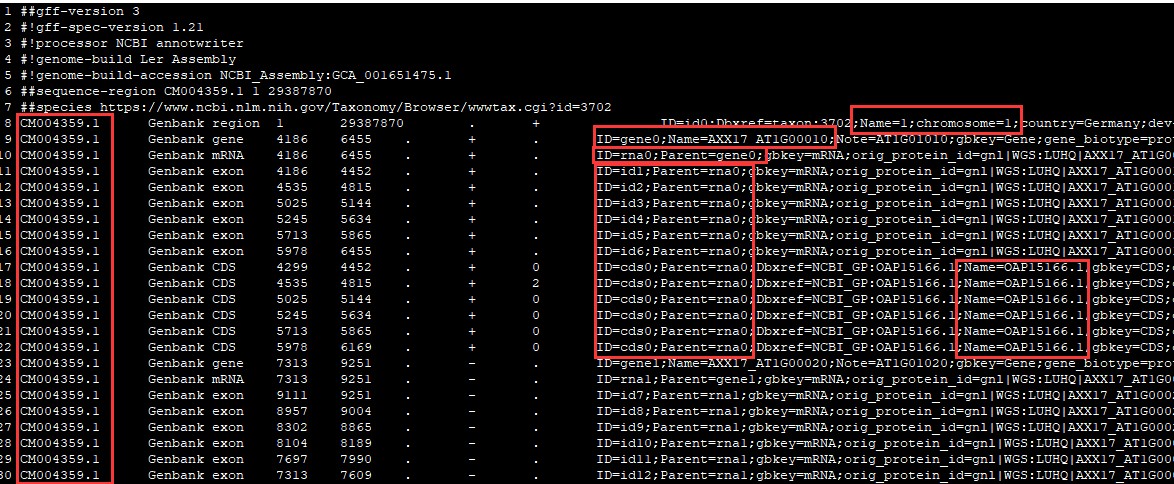
什么基因ID、染色体编号都是有自己命名规则的,跟我们通常用到的完全不一样。遇到这种情况小编都会去其他数据库下载基因组数据。但是,总会有部分基因组是在其他数据库里找不到的,那就只能认命用NCBI上的基因组了,但说实话真的不好用。
因此小编专门写了个perl脚本,将其改为正常命名的格式。修改后文件如下:
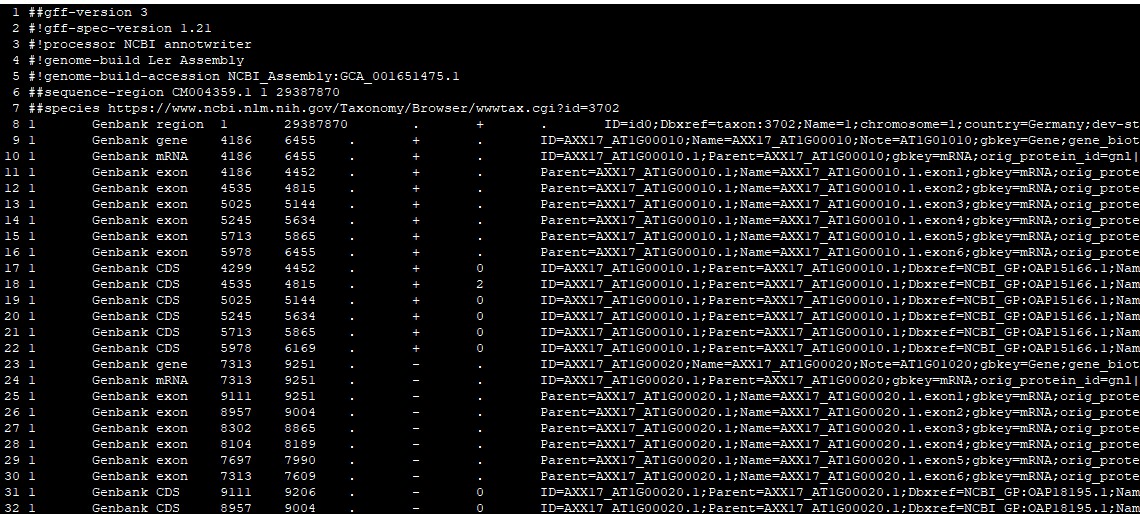
染色体编号、基因ID都改为正常的ID;转录本ID改为基因ID 加 ".1",如果有多个转录本,第二个转录本改为加 ".2",以此类推;CDS ID改为转录本ID。
脚本帮助
Usage
Forced parameter:
-gff genoma gff file <infile> must be given
-fa genoma fasta file <infile> must be given
-p protein fasta file <infile> optional
-pro_out output protein fasta file <outfile> optional
-o output genoma gff file <outfile> must be given
-out output genoma fasta file <outfile> must be given
Other parameter:
-h Help document
使用用法
运行命令如下:
perl ncbi_gff_2_Ensembl_gff.pl -gff genomic.gff -fa genomic.fa -o new.genomic.gff -out new.genomic.fna -p protein.fa -pro_out new.protein.fa
各参数作用:
-gff: 指定基因组gff文件
-fa: 指定基因组序列文件
-o: 指定新生成的基因组gff文件
-out: 指定新生成的基因组序列文件
-p: 指定基因组蛋白质序列文件
-pro_out: 指定新生成的基因组蛋白质序列文件
其中 -p为可选项,其后跟蛋白质序列文件,将蛋白质序列的序列ID改为转录本ID。如果有-p选项,则必须跟-pro_out选项,指定蛋白质序列ID替换后保存为的新文件。
由于NCBI中转录本序列的ID命名方式五花八门,所以没有通用的脚本可以修改其序列ID,只能具体情况具体对待,因此这里没有给出转录本序列文件修改的功能。
脚本代码
use Getopt::Long;
use strict;
use Bio::SeqIO;
use Bio::Seq;
#get opts
my %opts;
GetOptions(\%opts, "gff=s", "o=s", "fa=s", "p=s", "pro_out=s", "out=s","h");
if(!defined($opts{gff}) || !defined($opts{o}) || !defined($opts{fa}) || !defined($opts{out}) || defined($opts{h}))
{
print <<"Usage End.";
Usage
Forced parameter:
-gff genoma gff file <infile> must be given
-fa genoma fasta file <infile> must be given
-p protein fasta file <infile> optional
-pro_out output protein fasta file <outfile> optional
-o output genoma gff file <outfile> must be given
-out output genoma fasta file <outfile> must be given
Other parameter:
-h Help document
Usage End.
exit;
}
open(IN,"$opts{gff}") || die "open $opts{gff} failed\n";
open(OUT,">$opts{o}") || die "open $opts{o} failed\n";
my %chr;
my %gene;
my %gene_mrna_num;
my %mrna;
my %mrna_exon_num;
my %pro2mrna;
while(<IN>){
if(/^#/){
print OUT $_;
next;
}
chomp;
my @line = split("\t");
###########################################################################
if($line[2] eq "region"){
if($line[8] =~/chromosome/){
$line[8] =~ /chromosome=([^;]*)/;
my $chromosome = $1;
$chr{$line[0]} = $chromosome;
}else{
$chr{$line[0]} = $line[0];
}
}
###########################################################################
if($line[2] eq "gene"){
$line[8] =~ /ID=([^;]*);Name=([^;]*)/;
my $geneid = $1;
my $genename = $2;
$line[8] =~ s/$geneid/$genename/;
$gene{$geneid} = $genename;
$gene_mrna_num{$geneid} = 0;
}
###########################################################################
if($line[2] eq "mRNA"){
$line[8] =~ /ID=([^;]*);Parent=([^;]*)/;
my $mrnaid = $1;
my $geneid = $2;
$line[8] =~ s/$geneid/$gene{$geneid}/;
$gene_mrna_num{$geneid}++;
$line[8] =~ s/$mrnaid/$gene{$geneid}\.$gene_mrna_num{$geneid}/;
$mrna{$mrnaid} = "$gene{$geneid}.$gene_mrna_num{$geneid}";
$mrna_exon_num{$mrnaid} = 0;
}
###########################################################################
if($line[2] eq "exon"){
$line[8] =~ /ID=([^;]*);Parent=([^;]*)/;
my $exonid = $1;
my $mrnaid = $2;
$mrna_exon_num{$mrnaid}++;
$line[8] =~ s/$mrnaid/$mrna{$mrnaid};Name=$mrna{$mrnaid}\.exon$mrna_exon_num{$mrnaid}/;
$line[8] =~ s/ID=$exonid;//;
}
###########################################################################
if($line[2] eq "CDS"){
$line[8] =~ /ID=([^;]*);Parent=([^;]*)/;
my $cdsid = $1;
my $mrnaid = $2;
$line[8] =~ /Name=([^;]*)/;
my $proid = $1;
$pro2mrna{$proid} = $mrna{$mrnaid};
$line[8] =~ s/$mrnaid/$mrna{$mrnaid}/;
$line[8] =~ s/$cdsid/$mrna{$mrnaid}/;
}
$line[0] = $chr{$line[0]};
print OUT join("\t",@line)."\n";
}
close(IN);
close(OUT);
my $in = Bio::SeqIO->new(-file => "$opts{fa}" , -format => 'Fasta');
my $out = Bio::SeqIO->new(-file => ">$opts{out}" , -format => 'fasta');
while ( my $seq = $in->next_seq() ) {
my($id,$sequence,$desc)=($seq->id,$seq->seq,$seq->desc);
my $newSeqobj = Bio::Seq->new(-seq => $sequence,
-desc => $desc,
-id => $chr{$id},
);
$out->write_seq($newSeqobj);
}
if($opts{p}){
if(!defined($opts{pro_out}))
{
print "protein output file not defined\n";
exit;
}
my $in = Bio::SeqIO->new(-file => "$opts{p}" , -format => 'Fasta');
my $out = Bio::SeqIO->new(-file => ">$opts{pro_out}" , -format => 'fasta');
while ( my $seq = $in->next_seq() ) {
my($id,$sequence,$desc)=($seq->id,$seq->seq,$seq->desc);
my $newSeqobj = Bio::Seq->new(-seq => $sequence,
-desc => $desc,
-id => $pro2mrna{$id},
);
$out->write_seq($newSeqobj);
}
}
最后祝您科研愉快!
- 发表于 2018-11-16 15:39
- 阅读 ( 7623 )
- 分类:perl
