做微生物研究必懂的OTU table相关知识
微生物多样性分析中最基础、最重要的文件为OTU table,几乎所有的后续分析,如alpha多样性分析,beta多样性分析,差异分析等等都是基于OTU table展开的。因此理解OTU table的含义和来路,对微生物多样性分析至关重要。下面我们就来介绍一下OTU table是怎么样一步一步得来的,及其注意事项。
建库测序步骤:
16s/18S/ITS rRNA测序首先需要提取环境样品的DNA,这些DNA可以来自肠道、土壤、粪便、空气或水体等任何来源。
提取DNA后需要经过质检和纯化。
加入通用引物对16s/18S/ITS rRNA基因进行扩增,完成PCR扩增之后经过切胶回收,不同的样品再加上特定的测序接头,经过定量均匀混样之后,就可以直接上测序仪测序,得到原始测序reads;
illumina测序原理视频帮助理解;
https://v.qq.com/x/page/f0519pz5jmw.html
原始数据处理:
根据测序barcode序列区分不同的样本序列,将数据分开至不同的样品中。
原始测序数据需要去除接头序列,并将双端测序序列通过序列之间的overlap拼接成单条序列(Tags),此步可由flash[1]软件完成。
过滤低质量序列和去除嵌合体序列。
什么是嵌合体?
在PCR反应中,在延伸阶段由于不完全延伸,就会导致嵌合体序列的出现。如下图所示:在扩增序列Template1的过程中,在序列延伸阶段,只产生了部分Template1序列在延伸阶段就结束了,在下一轮的PCR反应中,这部分序列作为序列Template2的引物接着延伸,扩增就会形成Template1和Template2的嵌合体序列。通常在PCR过程中,大概有1%的几率会出现嵌合体序列,而在16S/18S/ITS 扩增子测序的分析中,由于序列相似度很高,嵌合体可达1%-20%,因此需要去除嵌合体序列。去除嵌合体可以将拼接好的Tags比对到参考数据库当中确定嵌合体,然后进行去除,这一步可以用mothur[2]软件实现。
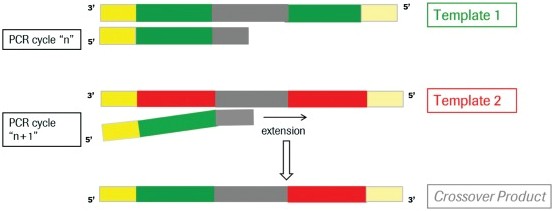 图注:PCR嵌合体生成
图注:PCR嵌合体生成
用vsearch去除嵌合体:
for i in `cat $fastmap |grep -v '#'|cut -f 1`; do
#相同重复序列合并
vsearch --derep_fulllength $workdir/3.data_qc/${i}.clean_tags.fq.gz \
--sizeout --output ${i}.derep.fa
#去嵌合体
vsearch --uchime3_deno ${i}.derep.fa \
--sizein --sizeout \
--nonchimeras ${i}.denovo.nonchimeras.rep.fa
#相同序列还原为多个
vsearch --rereplicate ${i}.denovo.nonchimeras.rep.fa --output ${i}.denovo.nonchimeras.fa
done
#根据参考序列去除嵌合体:
for i in `cat $fastmap |grep -v '#'|cut -f 1`; do
vsearch --uchime_ref ${i}.denovo.nonchimeras.fa \
--db $dbdir/rdp_gold.fa \
--sizein --sizeout --fasta_width 0 \
--nonchimeras ${i}.ref.nonchimeras.fa
done
什么是OTU
OTU(operational taxonomic units) 是在系统发生学研究或群体遗传学研究中,为了便于进行分析,人为给某一个分类单元(品系,种,属,分组等)设置的同一标志。通常按照 97% 的相似性阈值将序列划分为不同的 OTU,每一个 OTU 通常被视为一个微生物物种。相似性小于97%就可以认为属于不同的种,相似性小于95%,可以认为属于不同的属。
为什么引入OTU
高通量测序得到的16S序列有成千上万条,如果对每条序列都进行物种注释的话,工作量大、耗时长,而且16S扩增、测序等过程中出现的错误会降低结果的准确性。在16S分析中引入OTU,首先对相似性序列进行聚类,分成数量较少的分类单元,基于分类单元进行物种注释。这不仅简化工作量,提高分析效率,而且OTU在聚类过程中会去除一些测序错误的序列,提高分析的准确性。
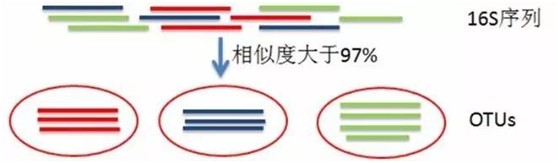
序列聚类形成OTU
聚类生成OTU的三种方法:
1、de novo OTU 聚类,是将所有序列直接按照两两之间的相似度,划分成一个个OTU,选取该OTU中丰度最高的序列作为该OTU的代表序列,然后用代表序列比对参考数据库,获得该OTU的物种注释。常用数据库有RDP、Silva及Greengene,由于GreenGene和RDP数据库一直没有更新,一般采用Silva数据库进行分析。
 OTU注释数据库
OTU注释数据库
优点:不依赖参考数据库,尤其是所研究的样品中含有的已知物种较少,如极端环境中。
缺点:受测序错误及嵌合体影响较大,说白了就是有些序列并非真实存在,是实验过程产生的“假序列”,用这种方法聚类时就会被误认为是一个独立的OTU,不过可以通过去嵌合体等分析手段缓解。
2、closed-reference聚类,这种方法是将序列与参考数据库直接比对,比对到同一参考序列的作为一个OTU,在OTU聚类的同时,也获得了该OTU的物种注释信息。
优点:所获得的OTU可信度高;另外,由于不同文章中检测的16S区域不同,如果要合并分析,不能用de novo OTU picking的方法聚类,因此只能用close-reference方法聚类。
缺点:只能得到已知物种的序列,丢失未知物种的信息。
3 、open-reference OTU聚类,具有上述两种聚类方法的特点,即将序列与参考序列比对,未比对上的序列再进行de novo聚类。兼具上述两种方法的优点,但无法用于不同16S区域的合并分析。
由于目前的参考数据库信息有限,所以OTU的注释结果中常见到一些uncultured*之类的没有分类信息。
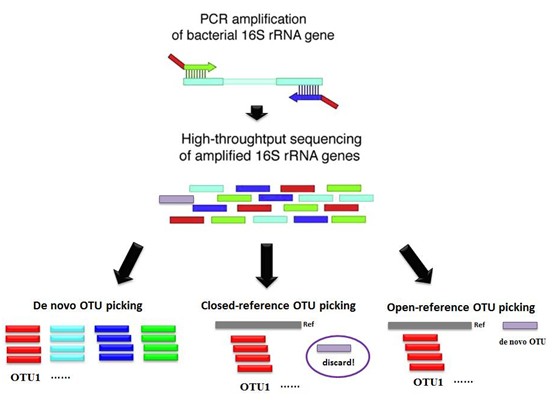
三种OTU聚类方法
经过OTU聚类和对OTU进行物种分类注释就可以得到一个OTU table了,这里包含每个样本所含的OTU种类及序列数,同时还有各个OTU的物种注释信息。一般如果没有特殊分析要求的话,应采用denovo的方法聚类获取OTU以最大限度的保留样品中物种种类,此分析过程可以用qiime[3]软件完成。具体如下图所示:
 OTU table
OTU table
如果某个样品的测序量较大(测序技术无法保证每个样品的测序量绝对的一致),相应的测到该样品中各种微生物的序列数会比其他样品多,即每个OTU中分到的序列数相应增加。因此OTU的序列数不能直接进行样品间的横向比较,而是要将序列数转化为比例也就是相对丰度(即序列数除以该样品的测序总序列数),得到该OTU在该样品中的比例,用这一比例进行横向比较。我们就可以说该OTU的丰度在不同样品间是升高了还是降低了。到这里,我们已经知道样品中的物种及其比例,可以根据比例绘制OTU丰度柱状图如下:
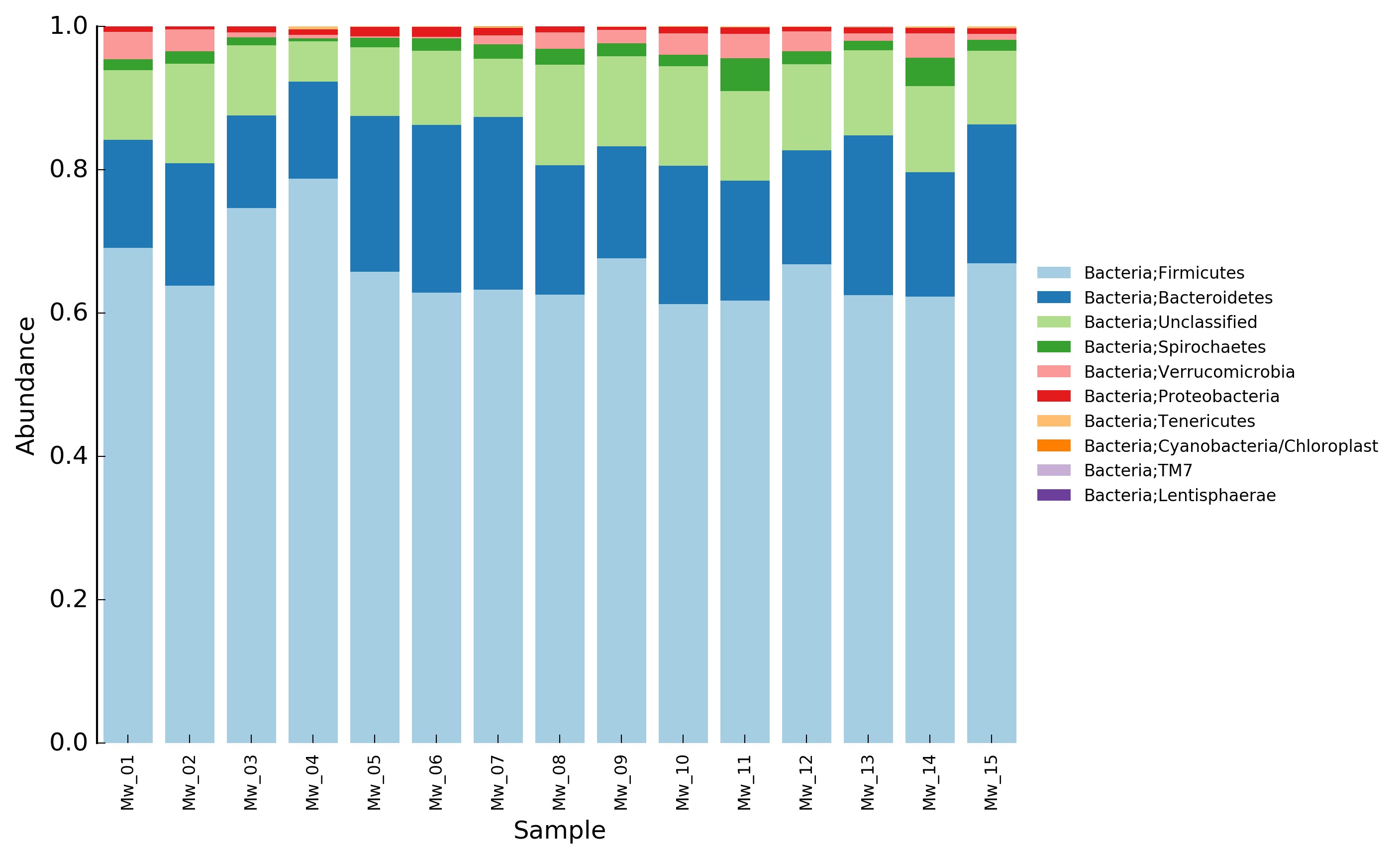 样品丰度比较
样品丰度比较
至此我们得到了,OTU table表格,后续的分析就很好展开了。
参考文献:
[1] T. Magoc and S. Salzberg. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27:21 (2011), 2957-63.
[2] Patrick D. Schloss,et al. Introducing mothur: Open-Source, Platform-Independent, Community-Supported Software for Describing and Comparing Microbial Communities. Appl. Environ. Microbiol. December 1, 2009 75:23 7537-7541;
[3] J Gregory Caporaso, et al. QIIME allows analysis of high-throughput community sequencing data. Nature Methods, 2010; doi:10.1038/nmeth.f.303
课程推荐:微生物扩增子分析课程实操 微生物16S/ITS/18S分析原理及结果解读
更多生物信息课程:
1. 文章越来越难发?是你没发现新思路,基因家族分析发2-4分文章简单快速,学习链接:基因家族分析实操课程、基因家族文献思路解读
2. 转录组数据理解不深入?图表看不懂?点击链接学习深入解读数据结果文件,学习链接:转录组(有参)结果解读;转录组(无参)结果解读
3. 转录组数据深入挖掘技能-WGCNA,提升你的文章档次,学习链接:WGCNA-加权基因共表达网络分析
4. 转录组数据怎么挖掘?学习链接:转录组标准分析后的数据挖掘、转录组文献解读
5. 微生物16S/ITS/18S分析原理及结果解读、OTU网络图绘制、cytoscape与网络图绘制课程
6. 生物信息入门到精通必修基础课:linux系统使用、biolinux搭建生物信息分析环境、linux命令处理生物大数据、perl入门到精通、perl语言高级、R语言画图、R语言快速入门与提高
7. 医学相关数据挖掘课程,不用做实验也能发文章:TCGA-差异基因分析、GEO芯片数据挖掘、 GEO芯片数据不同平台标准化 、GSEA富集分析课程、TCGA临床数据生存分析、TCGA-转录因子分析、TCGA-ceRNA调控网络分析
8.其他,二代测序转录组数据自主分析、NCBI数据上传、二代fastq测序数据解读、
9.组学大讲堂全部生物生信数据挖掘课程可点击:组学大讲堂视频课程
- 发表于 2018-04-20 19:53
- 阅读 ( 40121 )
- 分类:宏基因组
