SRA工具sratoolkit把原始测序数据转为fastq格式
SRA工具sratoolkit把原始测序数据转为fastq格式
我们下载好的SRA数据通常是下图这样的:
然后我们需要把当前文件夹下的所有sra都解压开来!
使用的命令非常简单:
$ fastq-dump SRR2090164.sra --split-3 -O ./ $ fastq-dump SRR2090164.sra --split-3 --gzip -O ./
解压的同时它也会显示每个SRA文件的数据量。
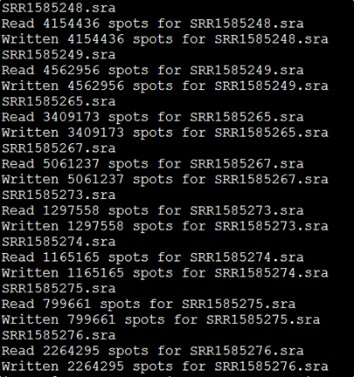
解压后文件如下:
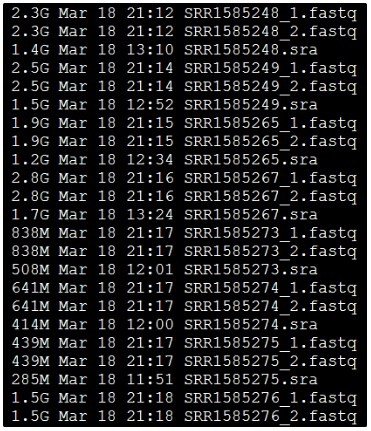
可以看到,每个SRA文件都产生了两个reads,分别是左右两端测序,说明这个SRA文件是双端测序策略。
此外,我们在网易云课堂上有各种教学视频,有兴趣可以了解一下:
1. 文章越来越难发?是你没发现新思路,基因家族分析发2-4分文章简单快速,学习链接:基因家族分析实操课程
2. 转录组数据理解不深入?图表看不懂?点击链接学习深入解读数据结果文件,学习链接:转录组(有参)结果解读;转录组(无参)结果解读
3. 转录组数据深入挖掘技能-WGCNA,提升你的文章档次,学习链接:WGCNA-加权基因共表达网络分析
4. 转录组数据怎么挖掘?学习链接:转录组标准分析后的数据挖掘
6. 更多学习内容:linux、perl、R语言画图,更多免费课程请点击以下链接:
- 发表于 2019-07-12 11:57
- 阅读 ( 6976 )
- 分类:软件工具
