GSEA分析需要的基因集Molecular Signatures Database是什么?
在作GSEA的分析时候需要提供一个预先定义好的基因集,用来评估基因集的基因在与表型相关度排序的基因表中的分布趋势,从而判断其对表型的影响作用,那么基因集都包括哪些呢? GSEA官网的基因集...
- 0
- 0
- landy
- 发布于 2018-05-31 16:03
- 阅读 ( 11749 )
aggregate对数据分组处理
利用aggregate对数据进行分组处理,包括分组求和,分组取均值,最大值,中位数等等
- 0
- 2
- Daitoue
- 发布于 2018-05-31 15:52
- 阅读 ( 9796 )
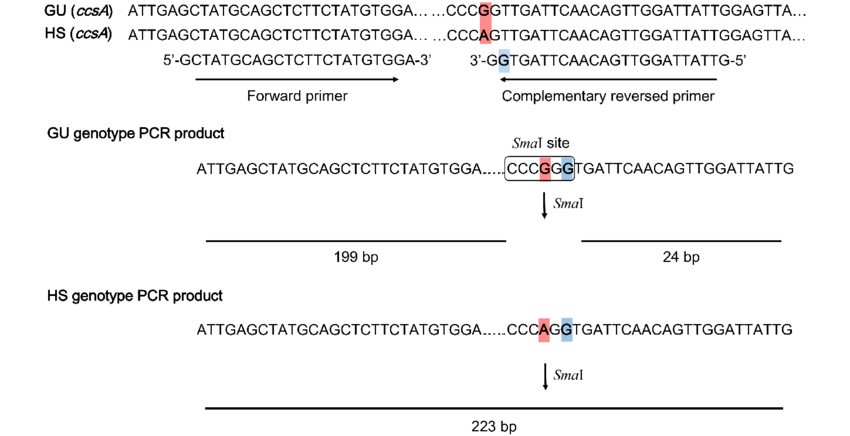
SNP不能转化成CAPS标记?来试试dCAPS吧!
dCAPS是由CAPS标记衍生的分子标记。它是通过引物引入错配的碱基,从而构建或者去除限制性内切酶识别位点的标记技术
- 2
- 8
- 安生水
- 发布于 2018-05-30 13:41
- 阅读 ( 23073 )
排序分析PCA、PCoA、CA、NMDS、RDA、CCA等区别与联系
降维排序分析方法包括:PCA、PCOA、CA、DCA、NMDS、RDA、CCA等等还有很多,理解他们的区别与联系才能熟练运用。
- 2
- 5
- omicsgene
- 发布于 2018-05-29 13:00
- 阅读 ( 46126 )
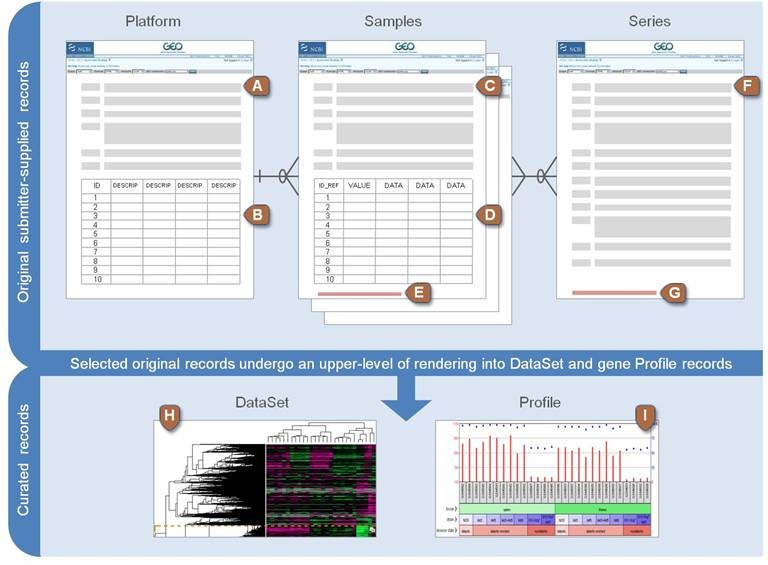
GEO数据库之芯片数据下载
教你利用GEOquery并结合Biobase下载GEO数据库多种数据,轻松数据下载与整理。
- 0
- 5
- Daitoue
- 发布于 2018-05-25 13:45
- 阅读 ( 13103 )
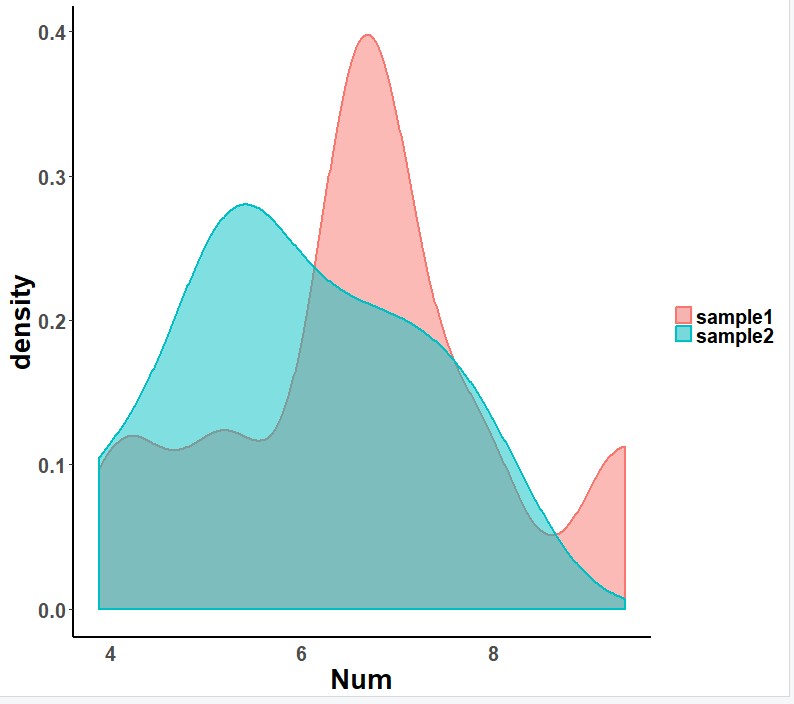

如何正确选择差异统计检验
对于科学问题,我们常常要找到差异,需要用到统计学检验,但是那么多统计学检验我该如何选择呢?
- 2
- 5
- omicsgene
- 发布于 2018-05-24 17:51
- 阅读 ( 14388 )
perl对文件夹处理
用perl批量处理文件夹下所有文件 opendir()获取目录权柄;readdir()读取目录下文件 #!/usr/bin/perl -w $dirname = "/tmp"; #指定一个目录 opendir ( DIR, $dirname ) || die...
- 1
- 4
- 安生水
- 发布于 2018-05-24 16:15
- 阅读 ( 3792 )
R语言计算平均值,中位数
Mean()求平均值 通过求出数据集的和再除以求和数的总量得到平均值 函数mean()用于在R语言中计算平均值。语法 用于计算R中的平均值的基本语法是 - mean(x, trim = 0, na.rm = FALSE, ...)...
- 1
- 2
- 安生水
- 发布于 2018-05-24 15:53
- 阅读 ( 14820 )
beta多样性中的距离矩阵总结
距离矩阵差别:jaccard bray-curtis 欧式距离 unifrac(weighted/unweigted)
- 1
- 4
- omicsgene
- 发布于 2018-05-24 13:20
- 阅读 ( 14094 )