如何快速获取多条序列长度信息
如何快速知道你的多个序列的长度呢?这里还是分享一个虚拟机的bio-linux的工具,samtools。命令行格式:samtools faidx xulie.fa(你的序列文件)结果会生成一个 xulie.fa.fai文件,也是就是在...
- 1
- 1
- landy
- 发布于 2018-05-11 11:31
- 阅读 ( 3563 )
Perl提取染色体序列指定范围的基因序列
Perl提取指定位置的基因序列用到 substr 函数,用法: $gene = substr( $fasta,$start-1, $end - $start+1); 其中$fasta是总的序列;$start是提取序列的开始位置,由于Perl字符串是从0计数的...
- 1
- 1
- 安生水
- 发布于 2018-05-09 16:27
- 阅读 ( 4256 )
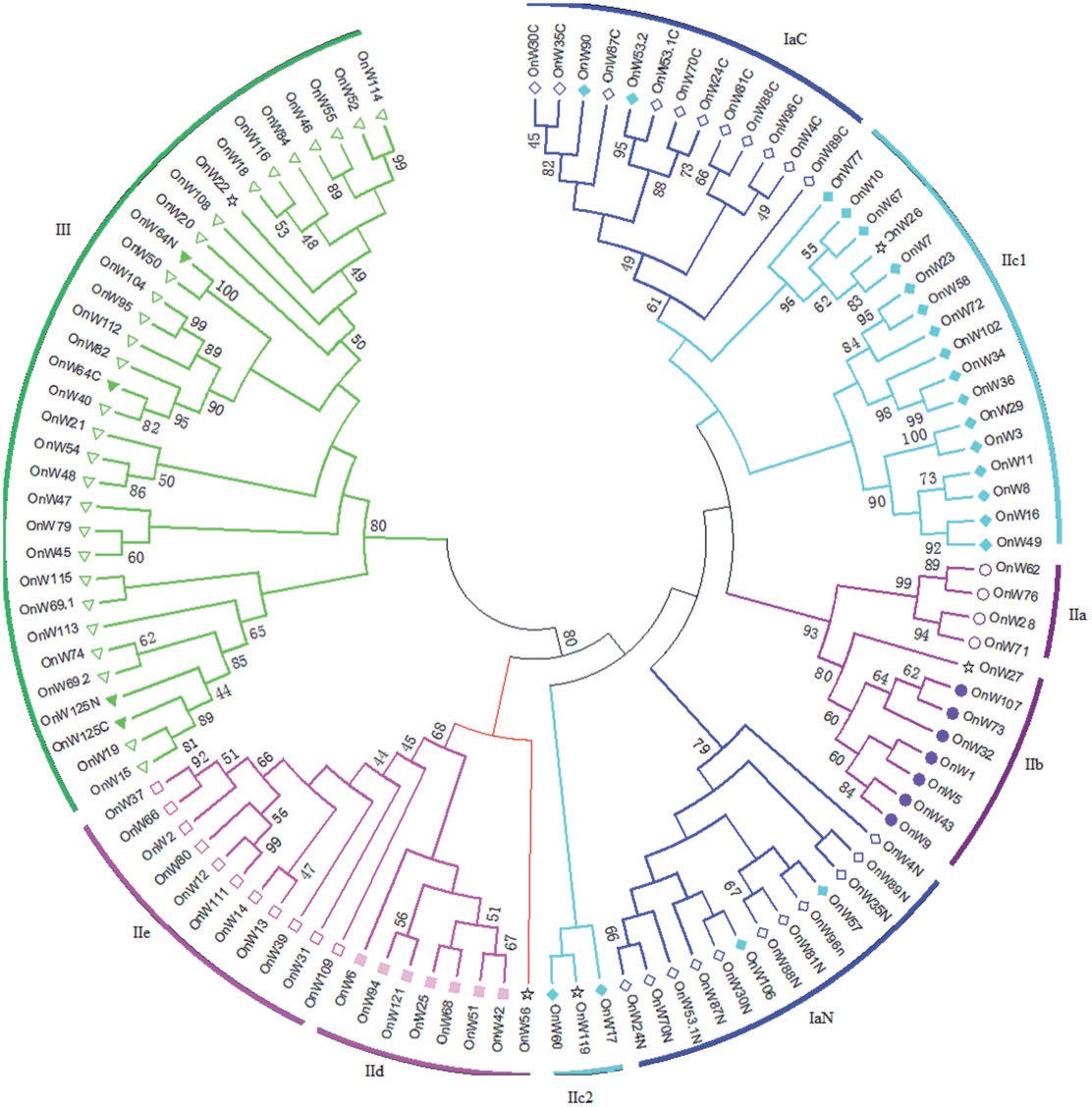
木薯NBS基因家族分析
利用公共数据发文章,基因家族好思路。 虽然我们一直在说可以“用别人的数据,写自己的文章”,但是具体怎么操作,很多同学还是不清楚。今天就用一个完全用公共数据库资源进行分析发文的例子来和大家分享下这方面的思路。 读前准...
- 1
- 1
- landy
- 发布于 2018-04-23 14:20
- 阅读 ( 8970 )
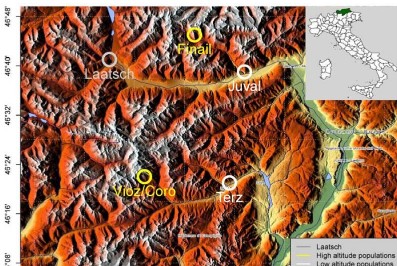

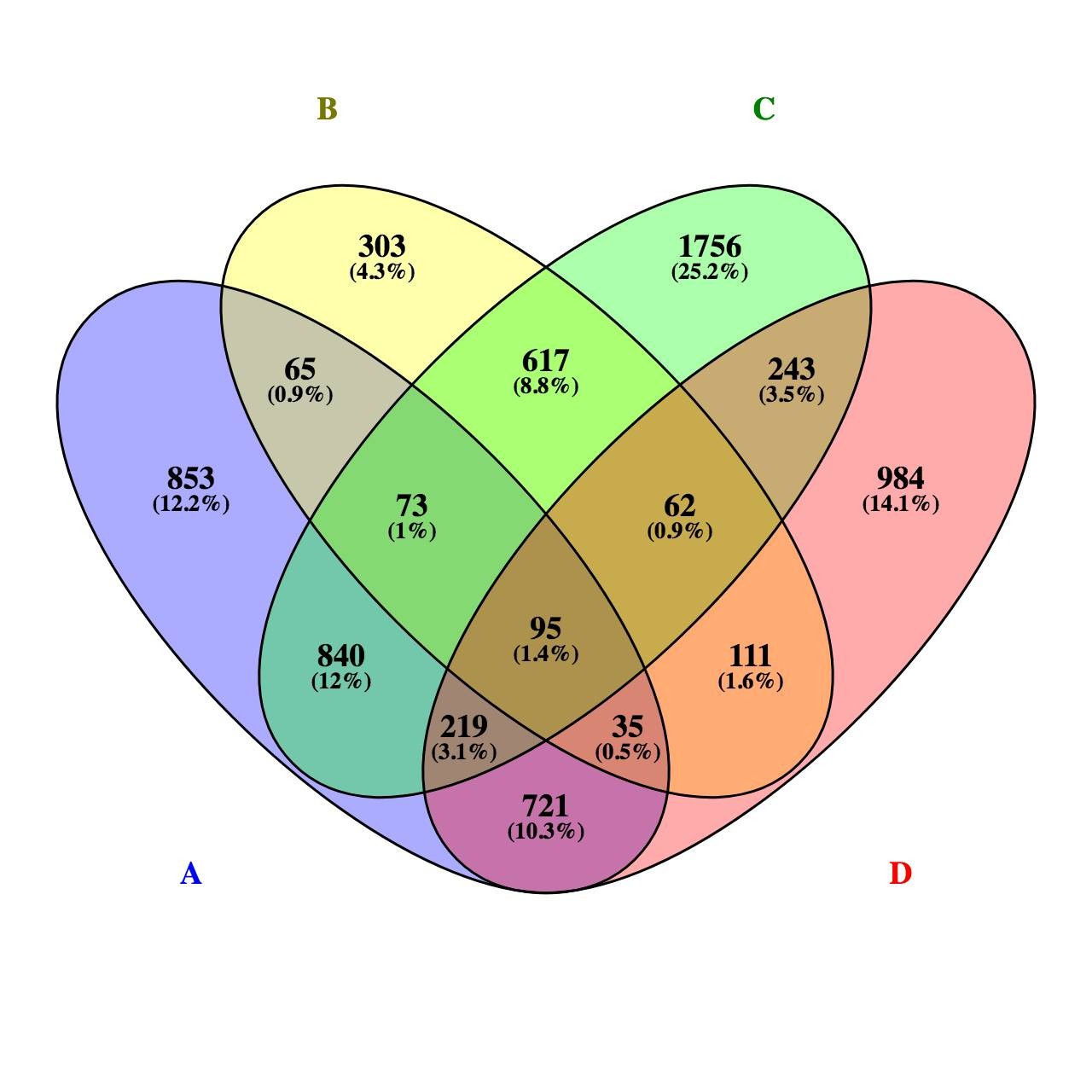

几个样品的转录组文章很难发了?这篇文章一定能给您启示
5个转录组也能发4分文章,想要少投入,多产出,既要站在巨人的肩膀上,也得打铁还需自身硬啊!
- 0
- 1
- omics007
- 发布于 2018-04-21 18:57
- 阅读 ( 12414 )


BaiduPCS-Go 报错 | 上传文件失败, 分片上传—合并分片文件: 遇到错误, 远端服务器返回错误, 代码: 31352, 消息: commit superfile2 failed
在使用BaiduPCS-Go上传文件时报错: [1] ↑ 1.03GB/1.03GB 1.81MB/s in 39s ............[1] 上传文件失败, 分片上传—合并分片文件: 遇到错误, 远端服务器返回错误, 代码: 31352, 消息: commit...
- 0
- 0
- rzx
- 发布于 4天前
- 阅读 ( 45 )
samtools | 提取部分染色体的bam信息
01. samtools提取bam文件中某部分染色体相关信息 samtools view -b -h XX.bam chr1 chr4 >XX2.bam 02. samtools构建bam文件的索引 samtools index XX2.bam
- 0
- 0
- rzx
- 发布于 4天前
- 阅读 ( 73 )
Trinity报错“Error, not recognizing read name formatting”
在使用trinity做de novo的时候,发现软件报错了
- 0
- 0
- 每天学习一点点
- 发布于 5天前
- 阅读 ( 54 )
hisat2-build报错:Error: Encountered internal HISAT2 exception (#1)
hisat2-build报错:Error: Encountered internal HISAT2 exception (#1)
- 0
- 0
- 安生水
- 发布于 2024-12-11 15:15
- 阅读 ( 99 )
文件夹名称中存在空格,scp如何正确识别
如果文件夹名称中有空格,如果不进行特殊处理scp指令无法对文件夹名称进行正确识别,就会出现“No such file or directory”的报错信息
- 0
- 0
- 每天学习一点点
- 发布于 2024-12-09 10:16
- 阅读 ( 123 )
IF=4.3 | 秋茄树SOS1基因家族分析
“ 2024年8月《BMC Plant Biology》发表了一篇题为“SOS1 gene family in mangrove (Kandelia obovata): Genome-wide identification, characterization, and expression analyses under salt and...
- 0
- 0
- rzx
- 发布于 2024-12-04 10:11
- 阅读 ( 167 )