R处理大数据时内存out of memory的问题
R处理大数据时偶尔会出现out of memory的问题,相关解决方法: 1、memory.size()查看当前work space内存使用状况(MB) 2、memory.limit()查看当前work space运行使用内存的上限 > memo...
- 0
- 0
- Daitoue
- 发布于 2018-06-29 13:16
- 阅读 ( 3959 )

你了解SAM和BAM文件吗
当我们测序得到的fastq文件map到基因组之后,会得到一个以SAM或Bam为扩展名的文件。这里将详细介绍SAM和Bam文件!
- 0
- 4
- 安生水
- 发布于 2018-06-29 09:33
- 阅读 ( 12626 )
统计不同样品-生物学重复基因表达量数据合并和绘图统计
处理统计RNA-seq 表达量数据,绘制柱状图,PCA散点图,还有不同样品表达基因维恩图。
- 1
- 0
- omicsgene
- 发布于 2018-06-28 23:52
- 阅读 ( 12672 )
微生物互作网络构建教程网站--Microbial association network construction tutorial
基于微生物16s测序数据中的OTU丰度矩阵可以进行微生物互作网络构建,从而探索微生物之间的互作关系。而网络的构建方法多种多样,下面给大家推荐一个网站,Microbial association network constr...
- 1
- 0
- Daitoue
- 发布于 2018-06-28 22:18
- 阅读 ( 5529 )
转录组分析时,影响差异基因数量的因素有哪些?
差异基因的筛选是基于统计学意义的,不能直观的通过两个数值的大小判断差异基因的是否。 首先:受测序深度的影响,有些样品的测序深度较深,可能导致该样品的readcount数值较高,做差...
- 0
- 0
- landy
- 发布于 2018-06-28 16:33
- 阅读 ( 10678 )
转录组差异基因为什么要进行聚类分析?
聚类分析用于判断差异基因在不同实验条件下的表达模式,将表达模式相同或相近的基因聚集成类,进而识别未知基因的功能或已知基因的未知功能,这些同类基因可能具有相似的功能,共同参...
- 0
- 2
- landy
- 发布于 2018-06-28 16:28
- 阅读 ( 16736 )
转录组测序样品间的相关性有何意义?如何计算?
样品间的相关性反应了样品间的相似程度,即不同处理或组织的样品在表达水平方面的相似度。相关系数越 接近1,样品间的相似度越高,样品间的差异基因也越少。生物学重复间的样品的相关系数...
- 0
- 3
- landy
- 发布于 2018-06-28 14:02
- 阅读 ( 13196 )
Cufflinks,Stringtie 合并转录本之后,如何筛选新转录本?
GTF文件中class code 注释转录本的类型
- 0
- 0
- microRNA
- 发布于 2018-06-28 09:51
- 阅读 ( 7501 )
endnote 插入文献 变成临时 大括号{} 不自动更新文献
endnote 文献插入出现大括号,不自动更新解决办法
- 1
- 19
- omicsgene
- 发布于 2018-06-27 17:53
- 阅读 ( 34712 )
GSEA法基因功能富集分析原理详解!
在组学大讲堂之前的一篇微信文章:关注的功能基因集在转录组结果中表现如何?中,跟大家介绍了GSEA(Gene Set Enrichment Analysis)及其分析结果。GSEA是一种基于基因集的富集分析方法,在对...
- 2
- 16
- landy
- 发布于 2018-06-25 18:16
- 阅读 ( 43125 )
搭建生信编程环境eclipse+perl+python+R(java环境)
搭建自己的生信编程环境eclipse+perl+python+R(java环境)
- 1
- 0
- omicsgene
- 发布于 2018-06-25 15:45
- 阅读 ( 5465 )

TCGA和GEO套路文章解说
利用GEO数据TCGA数据筛选疾病相关标志分子,miRNA,mRNA,lncRNA,circRNA等;
- 0
- 0
- omicsgene
- 发布于 2018-06-23 14:08
- 阅读 ( 8122 )
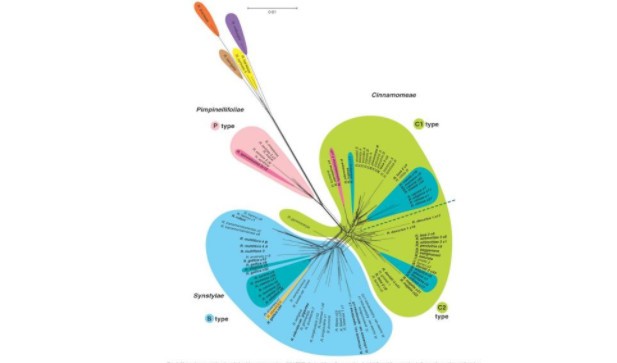
叶绿体基因做跨物种系统发育分析
叶绿体是绿色植物进行光合作用的细胞器,具有合成蛋白质、淀粉、色素等功能,普遍存在于真核自养生物中,尤其是藻类和陆生植物,其基因组可自主遗传。
- 1
- 0
- 安生水
- 发布于 2018-06-22 16:01
- 阅读 ( 5813 )
SNP2CAPS使用
SNP2CAPS可将SNP转换为CAPS标记,其具体用法: perl SNP2CAPS.pl chr5D:9950377.fa_1 link_gcg AanI,AarI,AasI,AatII,Aba6411II,AbaB8342IV,AbaCIII > chr5D:9950377.txt 其中...
- 1
- 0
- 安生水
- 发布于 2018-06-22 15:44
- 阅读 ( 4021 )