TCGA数据下载—TCGAbiolinks包参数详解
TCGA是由National Cancer Institute ( NCI, 美国国家癌症研究所) 和 National Human Genome Research Institute (NHGRI, 国家人类基因组研究所) 合作建立的癌症研究项目,通过收集整理癌症相关的各种组学数据,提供了一个大型的,免费的癌症研究参考数据库。 官方提供了对应的下载工具Genomic Data Commons Datga Portal, 简称GDC, 网址如下:https://portal.gdc.cancer.gov/,挖掘里面的数据发生信文章近年来非常火热,这里介绍一下利用R包TCGAbiolinks下
TCGAbiolinks包是从TCGA数据库官网接口下载数据的R包。它的一些函数能够轻松地帮我们下载数据和整理数据格式。
其实就是broad研究所的firehose命令行工具的R包装!
1.包的安装:
local({r <- getOption("repos")
r["CRAN"] <- "http://mirrors.tuna.tsinghua.edu.cn/CRAN/"
options(repos=r)})
if (!requireNamespace("BiocManager", quietly=TRUE)){
install.packages("BiocManager")
}
options(BioC_mirror="https://mirrors.tuna.tsinghua.edu.cn/bioconductor")
BiocManager::install("TCGAbiolinks")
library(TCGAbiolinks)
2.利用TCGAbiolinks下载数据
下载数据分为三步,分别用到TCGAbiolinks包中三个函数:
1)查询数据 GDCquery()
2)下载数据 getResults()
3)保存整理数据 GDCprepare()
以上三步中重点介绍第一个GDCquery()使用方法,其参数最多12个,而且每个参数可设置的选项也非常多,剩下两个函数,使用相对简单了。以下为使用方法和参数说明:
GDCquery(project, data.category, data.type, workflow.type,
legacy = FALSE, access, platform, file.type, barcode, data.format,
experimental.strategy, sample.type)
官方的参数说明比较简单:
简单的使用举例:
query <- GDCquery(project = "TCGA-ACC",
data.category = "Copy Number Variation",
data.type = "Copy Number Segment")
GDCquery参数说明:
1.project
可以通过getGDCprojects()$project_id,获取TCGA中最新的不同癌种的项目号,更新项目信息对应癌症名称:https://www.omicsclass.com/article/1061
> getGDCprojects()$project_id
[1] "TCGA-MESO" "TCGA-READ" "TCGA-SARC"
[4] "TCGA-ACC" "TCGA-LGG" "TCGA-THCA"
[7] "TARGET-CCSK" "TARGET-NBL" "BEATAML1.0-CRENOLANIB"
[10] "TARGET-AML" "TCGA-SKCM" "TCGA-CHOL"
[13] "TCGA-KIRC" "TCGA-BRCA" "VAREPOP-APOLLO"
[16] "HCMI-CMDC" "ORGANOID-PANCREATIC" "TCGA-GBM"
[19] "TCGA-OV" "FM-AD" "TCGA-UCEC"
[22] "TARGET-ALL-P3" "CGCI-BLGSP" "TARGET-ALL-P2"
[25] "TCGA-LAML" "TCGA-DLBC" "TCGA-KICH"
[28] "TCGA-THYM" "TCGA-UVM" "TCGA-PRAD"
[31] "TCGA-LUSC" "TCGA-TGCT" "CPTAC-3"
[34] "BEATAML1.0-COHORT" "TCGA-STAD" "TCGA-LIHC"
[37] "TCGA-COAD" "TARGET-OS" "TARGET-RT"
[40] "CTSP-DLBCL1" "TCGA-HNSC" "TCGA-ESCA"
[43] "TCGA-CESC" "TCGA-PCPG" "TCGA-KIRP"
[46] "TCGA-UCS" "TCGA-PAAD" "TCGA-LUAD"
[49] "TARGET-WT" "MMRF-COMMPASS" "TCGA-BLCA"
[52] "NCICCR-DLBCL" "TARGET-ALL-P1"
2.data.category
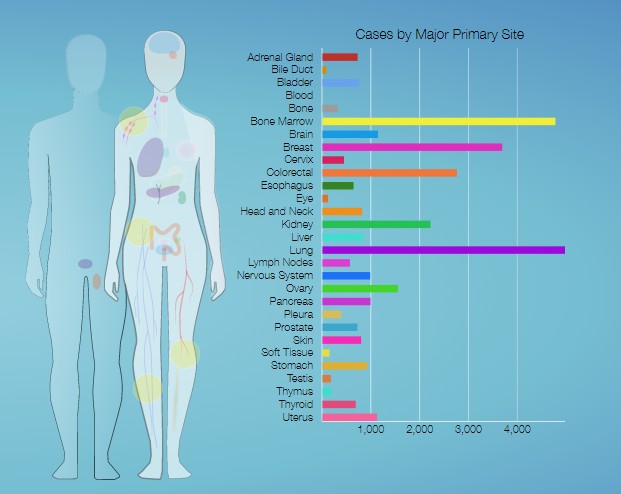
可以使用TCGAbiolinks:::getProjectSummary(project)查看project中有哪些数据类型,如查询"TCGA-ACC",有7种数据类型,case_count为病人数,file_count为对应的文件数。下载表达谱,可以设置data.category="Transcriptome Profiling":
> TCGAbiolinks:::getProjectSummary("TCGA-ACC")
$data_categories
case_count file_count data_category
1 80 397 Transcriptome Profiling
2 92 361 Copy Number Variation
3 92 744 Simple Nucleotide Variation
4 80 80 DNA Methylation
5 92 105 Clinical
6 92 352 Sequencing Reads
7 92 517 Biospecimen
$case_count
[1] 92
$file_count
[1] 2556
$file_size
[1] 3.920606e+12
3.data.type
这个参数受到上一个参数的影响,不同的data.category,会有不同的data.type,如下表所示:
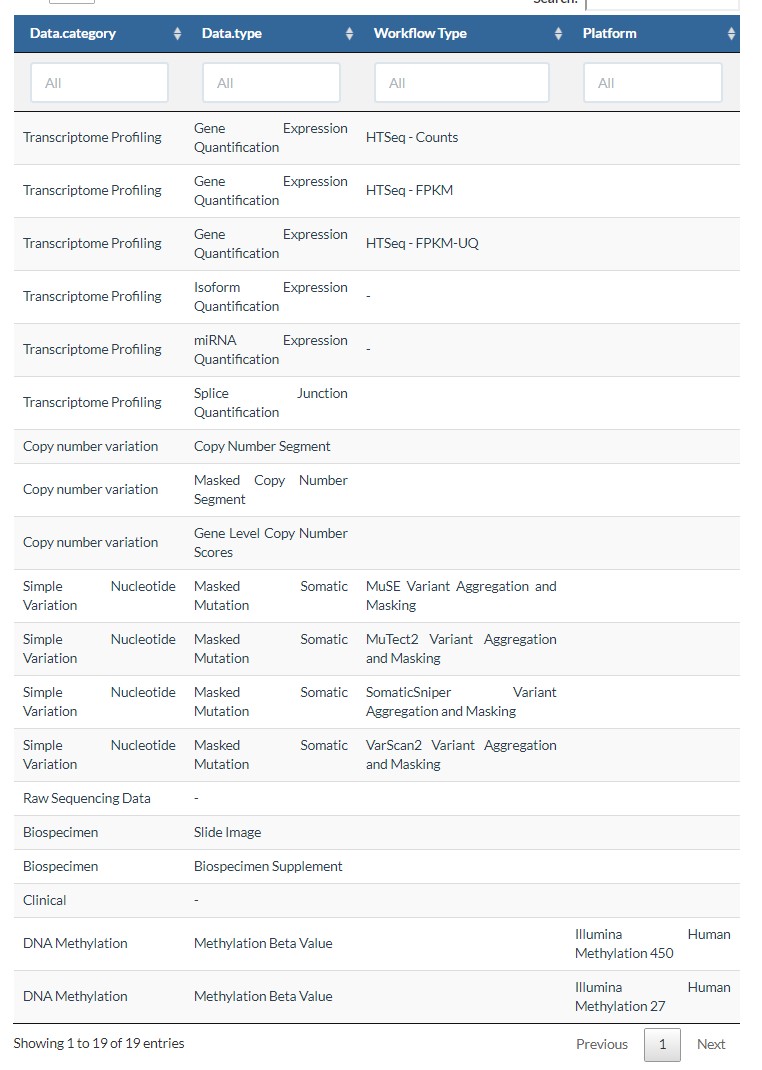
如果下载表达数据,常用的设置如下:
#下载rna-seq转录组的表达数据
data.type = "Gene Expresion Quantification"
#下载miRNA表达数据数据
data.type = "miRNA Expression Quantification"
#下载Copy Number Variation数据
data.type = "Copy Number Segment"
4.workflow.type
这个参数受到上两个参数的影响,不同的data.category和不同的data.type,会有不同的workflow.type,如下表所示:
如果下载表达数据,会有三种数据,FPKM,Counts,FPKM-UQ,区别见:https://www.omicsclass.com/article/1059
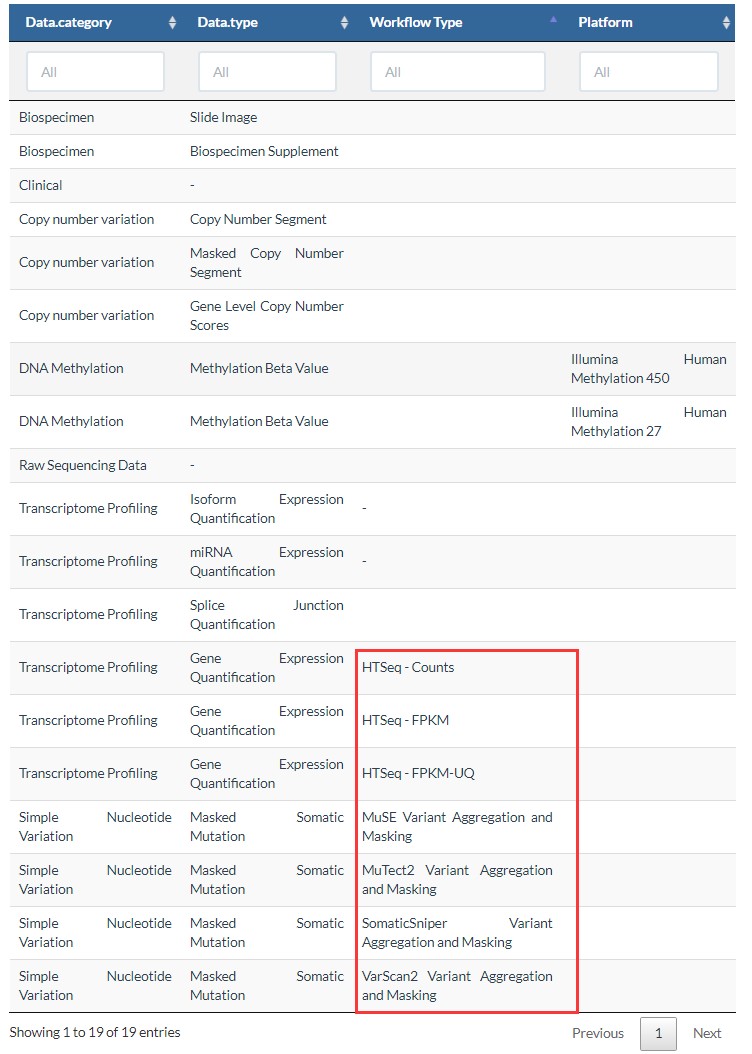
5 legacy
这个参数主要是设置TCGA数据有两不同入口可以下载,GDC Legacy Archive 和 GDC Data Portal,以下是官方的解释两种数据Legacy or Harmonized区别:大致意思为:Legacy 数据hg19和hg18为参考基因组(老数据)而且已经不再更新了,Harmonized数据以hg38为参考基因组的数据(新数据),现在一般选择Harmonized。
Different sources: Legacy vs Harmonized
There are two available sources to download GDC data using TCGAbiolinks:
- GDC Legacy Archive : provides access to an unmodified copy of data that was previously stored in CGHub and in the TCGA Data Portal hosted by the TCGA Data Coordinating Center (DCC), in which uses as references GRCh37 (hg19) and GRCh36 (hg18).
- GDC harmonized database: data available was harmonized against GRCh38 (hg38) using GDC Bioinformatics Pipelines which provides methods to the standardization of biospecimen and clinical data.
可以设置为TRUE或者FALSE:
Harmonized data options (legacy = FALSE)
Legacy archive data options (legacy = TRUE)
不同的的数据(新老Legacy or Harmonized),里面存储的数据会有差异,会影响前面data.category、 data.type 、 workflow.type参数的设置详细参考:http://www.bioconductor.org/packages/release/bioc/vignettes/TCGAbiolinks/inst/doc/query.html 这里贴一下如果是Harmonized data options (legacy = FALSE),前面三个参数可以设置的值如下:
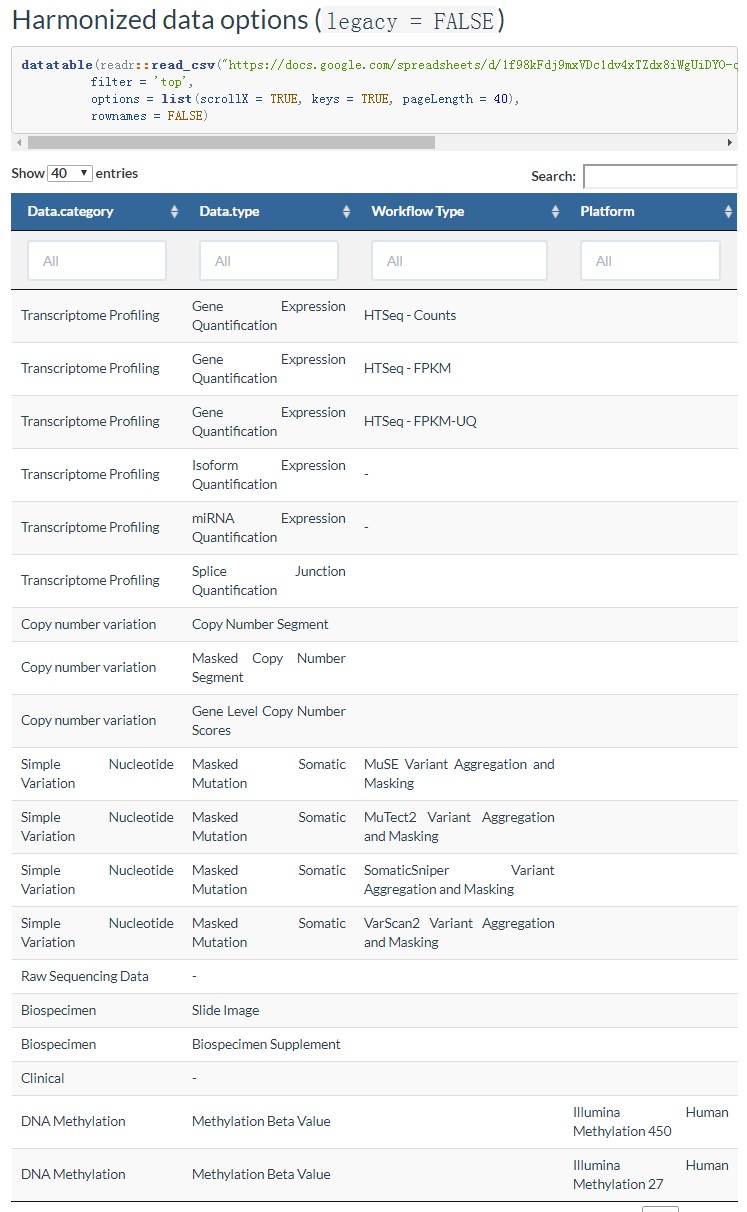 6 access
6 access
| Filter by access type. Possible values: controlled, open,筛选数据是否开放,这个一般不用设置,不开放的数据也没必要了,所以都设置成:access=“open" |
7.platform
涉及到数据来源的平台,如芯片数据,甲基化数据等等平台的筛选,一般不做设置,除非要筛选特定平台的数据:
| Example: | ||
| CGH- 1x1M_G4447A | IlluminaGA_RNASeqV2 | |
| AgilentG4502A_07 | IlluminaGA_mRNA_DGE | |
| Human1MDuo | HumanMethylation450 | |
| HG-CGH-415K_G4124A | IlluminaGA_miRNASeq | |
| HumanHap550 | IlluminaHiSeq_miRNASeq | |
| ABI | H-miRNA_8x15K | |
| HG-CGH-244A | SOLiD_DNASeq | |
| IlluminaDNAMethylation_OMA003_CPI | IlluminaGA_DNASeq_automated | |
| IlluminaDNAMethylation_OMA002_CPI | HG-U133_Plus_2 | |
| HuEx- 1_0-st-v2 | Mixed_DNASeq | |
| H-miRNA_8x15Kv2 | IlluminaGA_DNASeq_curated | |
| MDA_RPPA_Core | IlluminaHiSeq_TotalRNASeqV2 | |
| HT_HG-U133A | IlluminaHiSeq_DNASeq_automated | |
| diagnostic_images | microsat_i | |
| IlluminaHiSeq_RNASeq | SOLiD_DNASeq_curated | |
| IlluminaHiSeq_DNASeqC | Mixed_DNASeq_curated | |
| IlluminaGA_RNASeq | IlluminaGA_DNASeq_Cont_automated | |
| IlluminaGA_DNASeq | IlluminaHiSeq_WGBS | |
| pathology_reports | IlluminaHiSeq_DNASeq_Cont_automated | |
| Genome_Wide_SNP_6 | bio | |
| tissue_images | Mixed_DNASeq_automated | |
| HumanMethylation27 | Mixed_DNASeq_Cont_curated | |
| IlluminaHiSeq_RNASeqV2 | Mixed_DNASeq_Cont |
8 file.type
如果是在GDC Legacy Archive(legacy=TRUE)下载数据的时候使用,可以参考官网说明:http://www.bioconductor.org/packages/release/bioc/vignettes/TCGAbiolinks/inst/doc/query.html
如果在GDC Data Portal,这个参数不用设置
9 barcode
A list of barcodes to filter the files to download,可以指定要下载的样品,例如:
barcode =c"TCGA-14-0736-02A-01R-2005-01""TCGA-06-0211-02A-02R-2005-01"
10 data.format
可以设置的选项为不同格式的文件: ("VCF", "TXT", "BAM","SVS","BCR XML","BCR SSF XML", "TSV", "BCR Auxiliary XML", "BCR OMF XML", "BCR Biotab", "MAF", "BCR PPS XML", "XLSX"),通常情况下不用设置,默认就行;
11 experimental.strategy
用于过滤不同的实验方法得到的数据:
Harmonized: WXS, RNA-Seq, miRNA-Seq, Genotyping Array.
Legacy: WXS, RNA-Seq, miRNA-Seq, Genotyping Array, DNA-Seq, Methylation array, Protein expression array, WXS,CGH array, VALIDATION, Gene expression array,WGS, MSI-Mono-Dinucleotide Assay, miRNA expression array, Mixed strategies, AMPLICON, Exon array, Total RNA-Seq, Capillary sequencing, Bisulfite-Seq
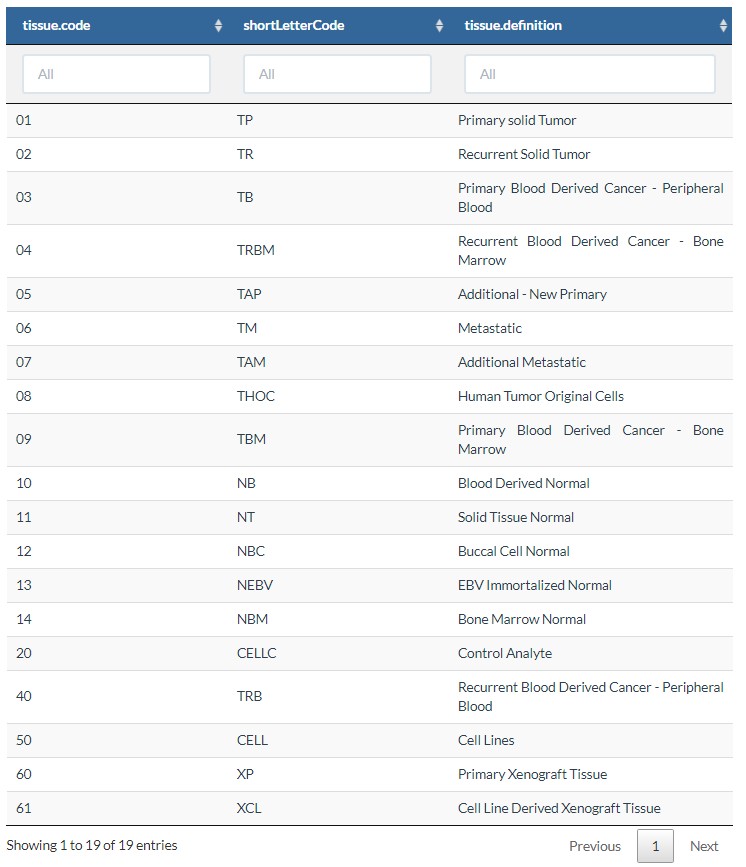
12 sample.type
对样本的类型进行过滤,例如,原发癌组织,复发癌等等;
学习完成了所有的参数,这里也有举例使用:
query <- GDCquery(project = "TCGA-ACC",
data.category = "Copy Number Variation",
data.type = "Copy Number Segment")
## Not run:
query <- GDCquery(project = "TARGET-AML",
data.category = "Transcriptome Profiling",
data.type = "miRNA Expression Quantification",
workflow.type = "BCGSC miRNA Profiling",
barcode = c("TARGET-20-PARUDL-03A-01R","TARGET-20-PASRRB-03A-01R"))
query <- GDCquery(project = "TARGET-AML",
data.category = "Transcriptome Profiling",
data.type = "Gene Expression Quantification",
workflow.type = "HTSeq - Counts",
barcode = c("TARGET-20-PADZCG-04A-01R","TARGET-20-PARJCR-09A-01R"))
query <- GDCquery(project = "TCGA-ACC",
data.category = "Copy Number Variation",
data.type = "Masked Copy Number Segment",
sample.type = c("Primary solid Tumor"))
query.met <- GDCquery(project = c("TCGA-GBM","TCGA-LGG"),
legacy = TRUE,
data.category = "DNA methylation",
platform = "Illumina Human Methylation 450")
query <- GDCquery(project = "TCGA-ACC",
data.category = "Copy number variation",
legacy = TRUE,
file.type = "hg19.seg",
barcode = c("TCGA-OR-A5LR-01A-11D-A29H-01"))
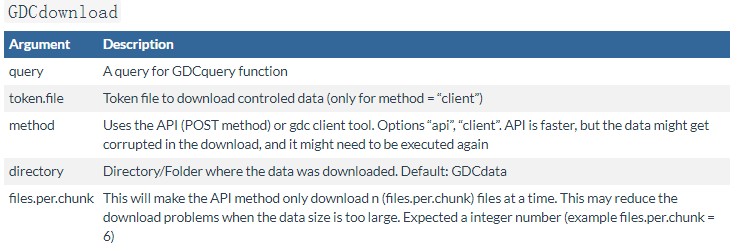
下载数据 GDCdownload()
上面的GDCquery()命令完成之后我们就可以用GDCdownload()函数下载数据了,如果数据很多,如果中间中断可以重复运行GDCdownload()函数继续下载,直到所有的数据下载完成,使用举例如下:
query <-GDCquery(project = "TCGA-GBM", data.category = "Gene expression", data.type = "Gene expression quantification", platform = "Illumina HiSeq", file.type = "normalized_results", experimental.strategy = "RNA-Seq", barcode = c("TCGA-14-0736-02A-01R-2005-01", "TCGA-06-0211-02A-02R-2005-01"), legacy = TRUE) GDCdownload(query, method = "client", files.per.chunk = 10, directory="D:/data")
具体参数说明如下,主要设置的参数:
- method如果设置为client 需要将gdc-client软件所在的路径添加到环境变量中,参考:gdc-client下载TCGA数据;
- query,为GDCquery查询的结果,
- files.per.chunk = 10,设置同时下载的数量,如果网速慢建议设置的小一些,
- directory="D:/data" 数据存储的路径;
整理数据 GDCprepare()
GDCprepare可以自动的帮我们获得基因表达数据:
data <- GDCprepare(query = query,
save = TRUE,
directory = "D:/data", #注意和GDCdownload设置的路径一致GDCprepare才可以找到下载的数据然后去处理。
save.filename = "GBM.RData") #存储一下,方便下载直接读取
获得了data数据之后,就可以往下数据挖掘了;
延伸阅读
更多生物信息课程:
1. 文章越来越难发?是你没发现新思路,基因家族分析发2-4分文章简单快速,学习链接:基因家族分析实操课程、基因家族文献思路解读
2. 转录组数据理解不深入?图表看不懂?点击链接学习深入解读数据结果文件,学习链接:转录组(有参)结果解读;转录组(无参)结果解读
3. 转录组数据深入挖掘技能-WGCNA,提升你的文章档次,学习链接:WGCNA-加权基因共表达网络分析
4. 转录组数据怎么挖掘?学习链接:转录组标准分析后的数据挖掘、转录组文献解读
5. 微生物16S/ITS/18S分析原理及结果解读、OTU网络图绘制、cytoscape与网络图绘制课程
6. 生物信息入门到精通必修基础课:linux系统使用、biolinux搭建生物信息分析环境、linux命令处理生物大数据、perl入门到精通、perl语言高级、R语言画图、R语言快速入门与提高、python语言入门到精通
7. 医学相关数据挖掘课程,不用做实验也能发文章:TCGA-差异基因分析、GEO芯片数据挖掘、 GEO芯片数据不同平台标准化 、GSEA富集分析课程、TCGA临床数据生存分析、TCGA-转录因子分析、TCGA-ceRNA调控网络分析
8.其他,二代测序转录组数据自主分析、NCBI数据上传、二代fastq测序数据解读、
9.全部课程可点击:组学大讲堂视频课程
- 发表于 2019-10-17 13:55
- 阅读 ( 26929 )
- 分类:TCGA
