GEO数据库挖掘—WGCNA鉴定骨肉瘤转移相关基因
骨肉瘤又称为成骨肉瘤,是一种恶性骨肿瘤,多出现早期转移和高死亡率。更好的研究和探索骨肉瘤早期转移的分子机制将有利于进行癌症的治疗,刚好这里就给大家介绍一篇利用公共数据分析的文献:基于共表达分析鉴定骨肉瘤转移相关基因。
数据来源
进行数据挖掘分析的所有数据都来自GEO数据库,数据下载基于两个原则:数据集属于骨肉瘤基因表达谱数据;数据集要求包含骨肉瘤转移和未转移这一分类信息。共获得四个数据集:GSE14359、GSE21257、 GSE32981、GSE14827。
数据分析
1.数据预处理
从GEO数据库下载四个数据集对应的芯片平台不同,将按照不同的方法进行处理。GSE14359和GSE14827对应的原始数据为CEL 文件格式,将基于Affy包进行背景校正和标准化。GSE21257、 GSE32981下载的原始数据为txt文件格式,基于limma包完成了校正工作。处理后的四个数据集数据将进行后续分析,其中GSE14359主要进行差异表达分析和WGCNA挖掘关键模块,剩下的三个数据集参与验证和分类分析。
2.差异表达分析
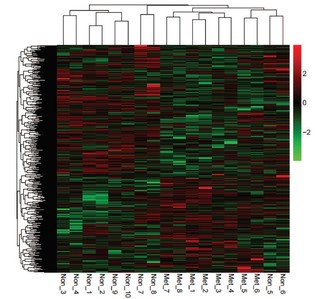
基于GSE14359表达谱数据进行差异分析,利用R语言limma包完成差异表达基因(DEG)的筛选,筛选标准为FDR<0.05 & |logFC|>1,最终获得了897 DEGs,并对筛选到的DEGs进行了双向层次聚类(见下图),在转移和未转移的样品中,基因表达差异趋势明显。
3.加权基因共表达网络分析
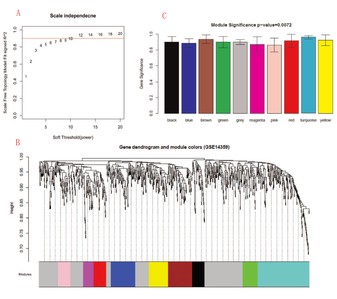
基于上一步分析筛选获取的差异基因进行WGCNA分析,power值选定12(R2>0.9,见下图A),模块最小基因数量设置大于30,并设定模块合并剪切高度0.95,共获得9个有效模块(含grey为10,见下图B),同时结合性状数据分析各模块基因与性状之间的GS(gene significance),并统计各模块内平均的GS作为MS(module significance)从而多方面衡量和选择重要模块进一步分析(见下图C)。
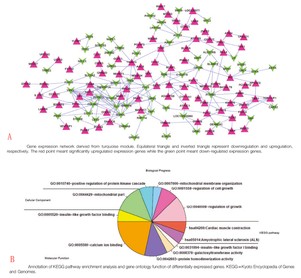
最终选定了模块基因数量最多(142),平均GS最高的Turquoise模块,构建共表达网络(见下图A),并进行GO、KEGG功能注释(见下图B)。该模块内上调基因83,下调基因59,进行功能注释的结果显示这些基因涉及的胰岛素样生长因子结合等功能可能和癌细胞转移进程有关。
4.SVM验证和评估
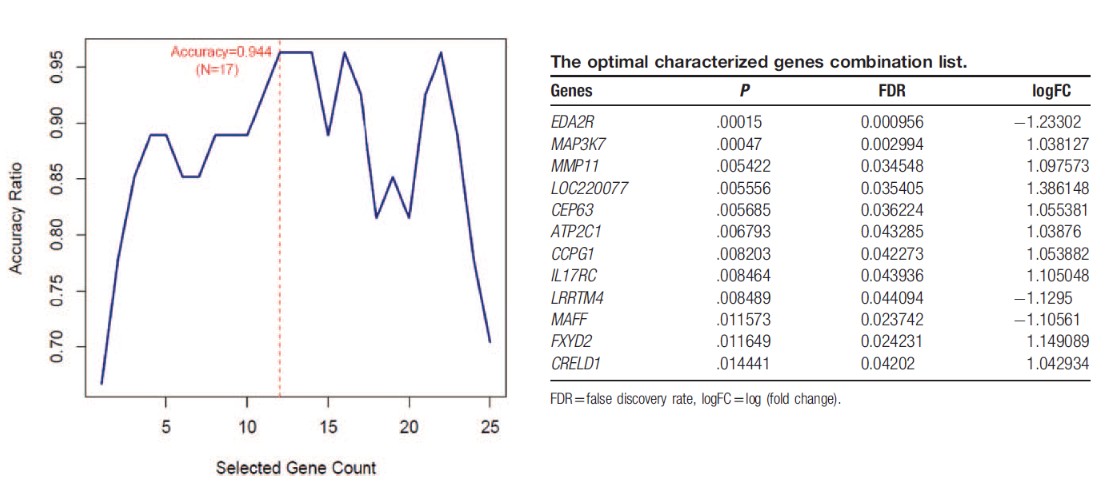
采用支持向量机(SVM)进行数据验证。基于GSE14359筛选分析获得142个基因,为进一步进行确认,选择该数据集作为训练数据,最终确定12个基因,对该17个样本可以进行准确的分类(accuracy rate of 94.4%,见下图),GSE21257、 GSE32981、GSE14827数据进行验证,GSE14827准确率96.3%,GSE32981准确率95.7%,GSE21257准确率92.3%。
结论
基于筛选分析以及对筛选基因的功能注释结果表明涉及胰岛素样生长因子结合蛋白IGFBPs可能和癌细胞的转移有关,同时SVM验证筛选的结果中MMP11基因等也可能和相关的癌细胞转移有关,也有实验表明口腔鳞状细胞癌细胞的转移过程中往往出现过表达等现象。
参考文献:
Tian H , Guan D , Li J . Identifying osteosarcoma metastasis associated genes by weighted gene co-expression network analysis (WGCNA)[J]. Medicine, 2018, 97(5):1966.
相关课程:
TCGA-基因差异表达分析、 TCGA-生存分析 、 GEO芯片数据挖掘、GEO芯片数据标准化、WGCNA-加权基因共表达网络分析
更多生物信息课程:
1. 文章越来越难发?是你没发现新思路,基因家族分析发2-4分文章简单快速,学习链接:基因家族分析实操课程、基因家族文献思路解读
2. 转录组数据理解不深入?图表看不懂?点击链接学习深入解读数据结果文件,学习链接:转录组(有参)结果解读;转录组(无参)结果解读
3. 转录组数据深入挖掘技能-WGCNA,提升你的文章档次,学习链接:WGCNA-加权基因共表达网络分析
4. 转录组数据怎么挖掘?学习链接:转录组标准分析后的数据挖掘、转录组文献解读
5. 微生物16S/ITS/18S分析原理及结果解读、OTU网络图绘制、cytoscape与网络图绘制课程
6. 生物信息入门到精通必修基础课:linux系统使用、perl入门到精通、perl语言高级、R语言入门、R语言画图
7. 医学相关数据挖掘课程,不用做实验也能发文章:TCGA-差异基因分析、GEO芯片数据挖掘、GEO芯片数据标准化、GSEA富集分析课程、TCGA临床数据生存分析、TCGA-转录因子分析、TCGA-ceRNA调控网络分析
8.其他,二代测序转录组数据自主分析、NCBI数据上传、二代测序数据解读
- 发表于 2019-10-25 11:38
- 阅读 ( 6850 )
- 分类:GEO
