使用Mfuzz进行转录组基因表达模式聚类分析
使用Mfuzz进行转录组基因表达模式聚类分析
Mfuzz是用来进行不同时间点转录组数据表达模式聚类分析的R包,使用起来非常方便,直接输入不同样本归一化后的counts或者FPKM及TPM值就可进行聚类。输入文件的格式很简单:
输入数据:
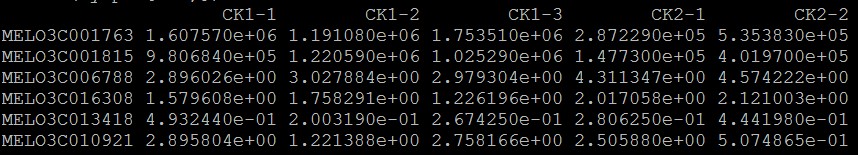
行为基因,列为样本,保存为制表符分隔的txt文件。
## 安装R包
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("Mfuzz")
## 加载R包
library("Mfuzz")
## 导入基因表达量
gene <- read.table("input.txt",header = T,row.names=1,sep="\t")
## 转换格式
gene_tpm <- data.matrix(gene)
eset <- new("ExpressionSet",exprs = gene_tpm)
## 过滤缺失超过25%的基因
gene.r <- filter.NA(eset, thres=0.25)由于输入的表达量中不允许有缺失值NA出现,所以我们要填补缺失值。
## mean填补缺失 gene.f <- fill.NA(gene.r,mode="mean") ## knn/wknn方法表现更好,但是计算起来比较复杂 gene.f <- fill.NA(gene.r,mode="knn") gene.f <- fill.NA(gene.r,mode="wknn") ## 过滤标准差为0的基因 tmp <- filter.std(gene.f,min.std=0) ## 标准化 gene.s <- standardise(tmp)
聚类时,我们需要输入两个参数,c和m。c是我们期望的聚类个数,由自己定义。m由mestimate计算得到。
## 聚类个数 c <- 6 ## 计算最佳的m值 m <- mestimate(gene.s) ## 聚类 cl <- mfuzz(gene.s, c = c, m = m) ## 查看每类基因数目 cl$size ## 查看每类基因ID cl$cluster[cl$cluster == 1] ## 输出基因ID write.table(cl$cluster,"output.txt",quote=F,row.names=T,col.names=F,sep="\t") ## 绘制折线图 mfuzz.plot(gene.s,cl,mfrow=c(2,3),new.window= FALSE)
更多生物信息课程:https://study.omicsclass.com/index
- 发表于 2020-07-01 10:03
- 阅读 ( 6993 )
- 分类:转录组
