IGV可视化使用
IGV是什么
Integrative Genomics Viewer(IGV) 是一款针对高通量测序数据进行可视化的专业软件。如转录组测序Reads与参考基因组序列比对结果,物种参考基因组序列和注释文件,都可使用IGV进行可视化浏览。 它支持各种数据类型,包括基于芯片测序、二代测序和基因组注释数据等。
IGV如何使用
一. 下载安装IGV
1.首先确保系统中已经安装好 Java JDK;正确配置好环境变量;
Java JDK下载地址:https://www.oracle.com/java/technologies/javase/javase-jdk8-downloads.html
环境变量配置帮助:https://www.omicsclass.com/article/298
2.下载安装IGV,打开网址:https://software.broadinstitute.org/software/igv/download
找到Windows版本,点击下载
下载完成
点击安装
安装完成,点开会先出现命令界面,再等一下会出现可视化界面。
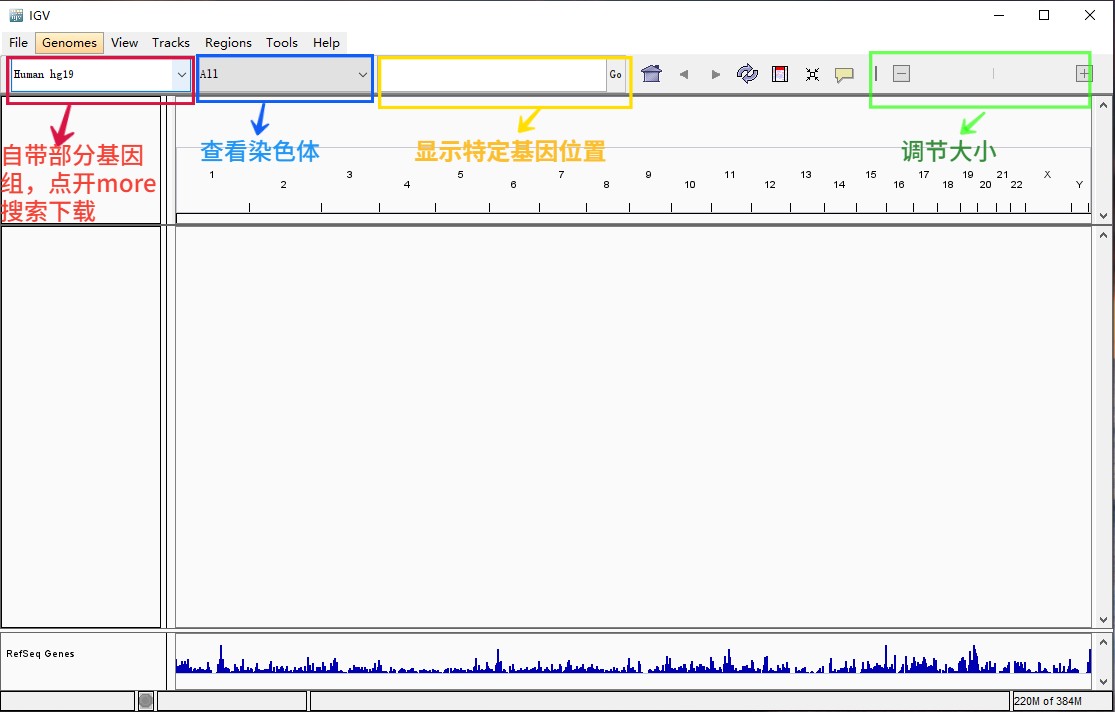
二. 加载参考基因组
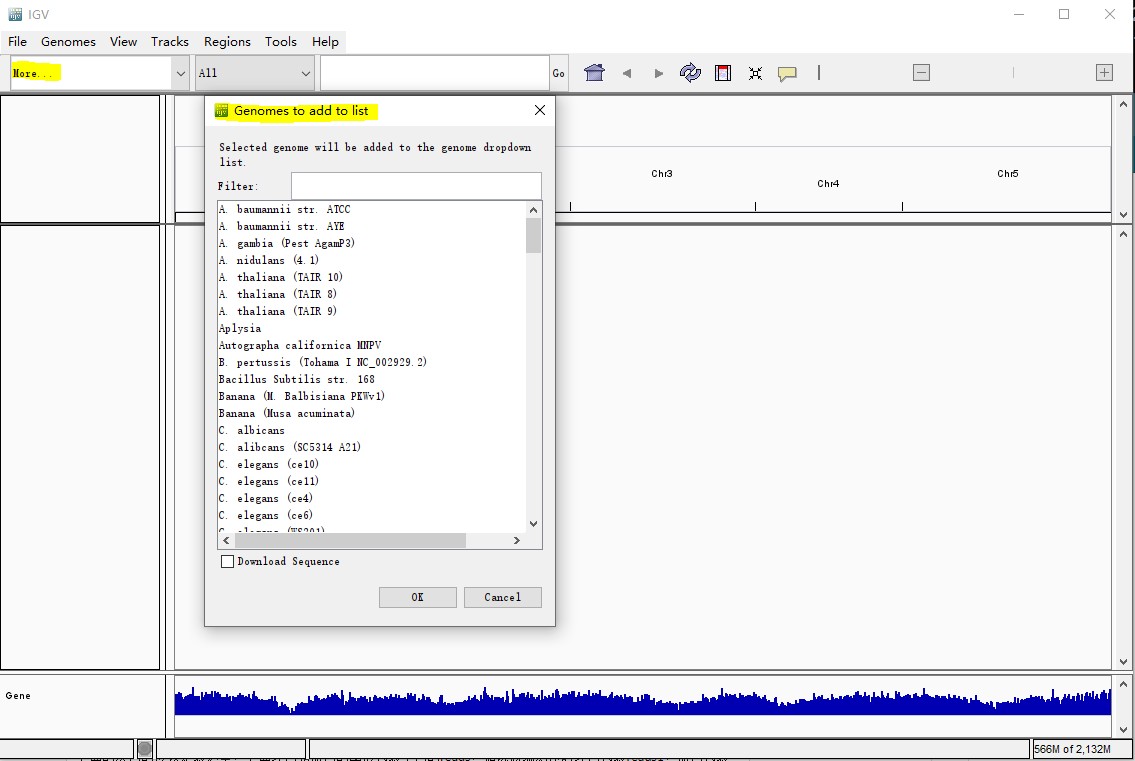
1.可以先点开上图红色框中的more选项,会出现软件自带的基因组,如果有需要的版本,直接点击下载即可。注意选择和测序数据一致的基因组,否则可能出错。
2.如果里面没有需要的基因组,可以自己构建基因组信息
下面以水稻基因组为例介绍如何使用:
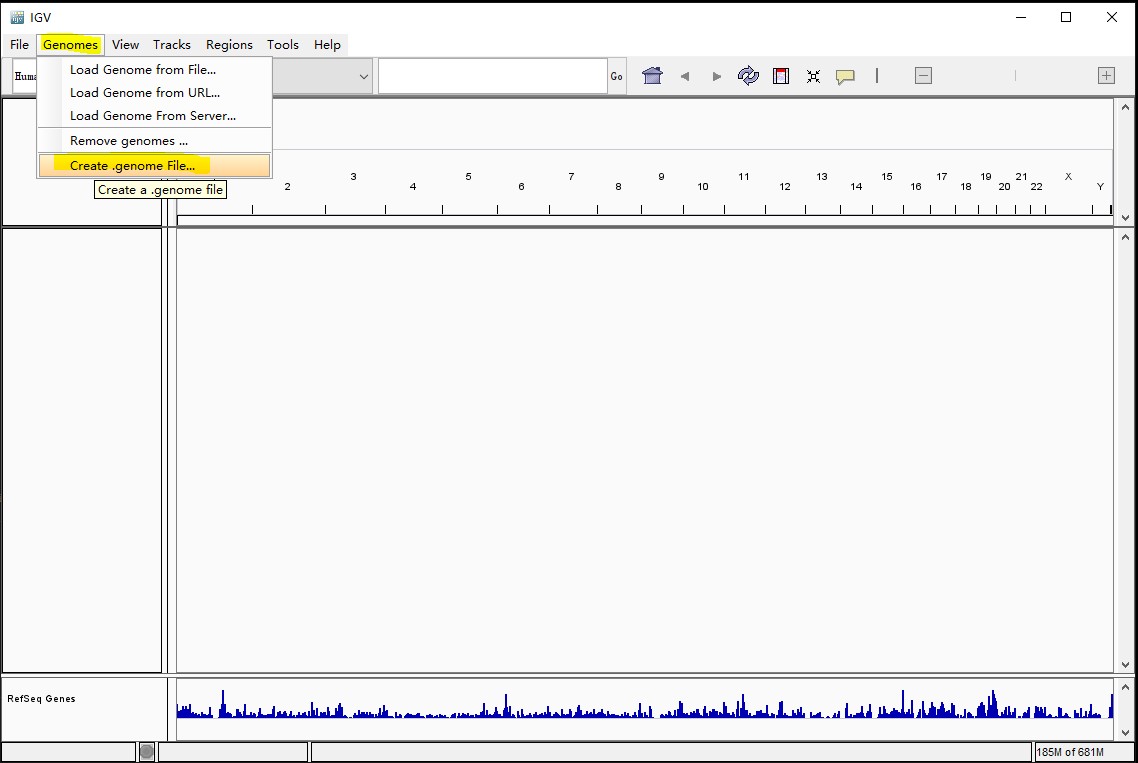
首先点击Genomes选项,选择Create .genome.File
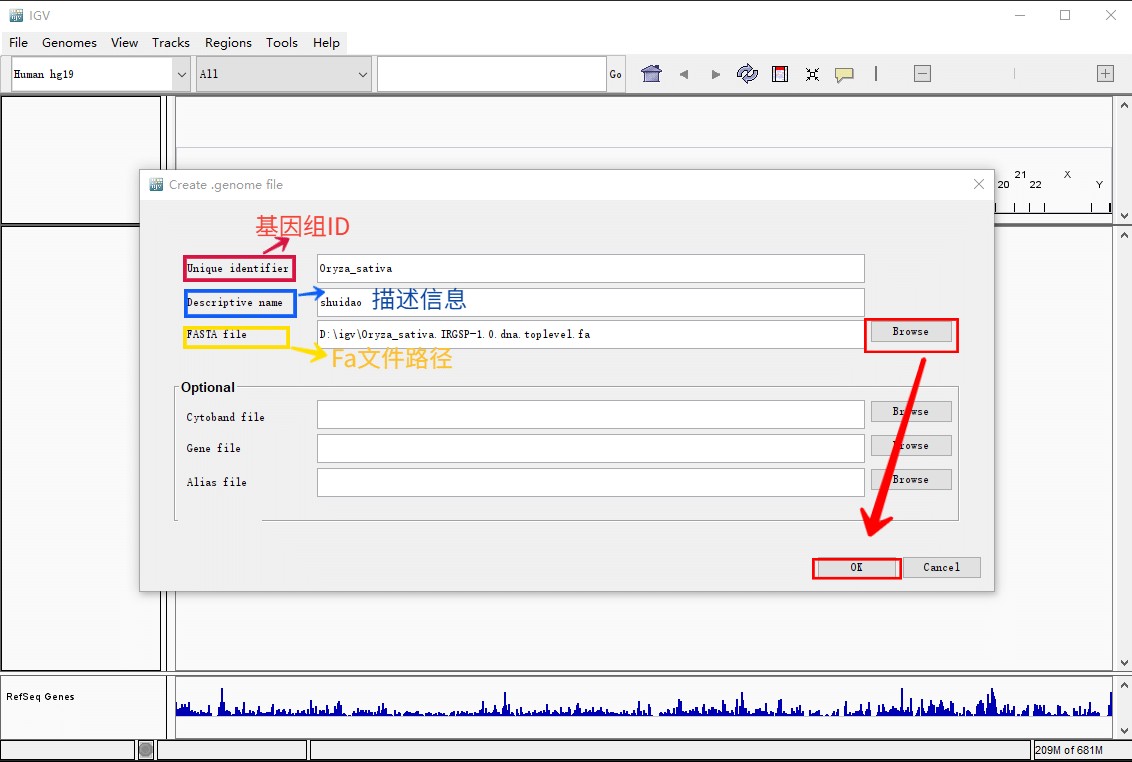
打开之后,按照下图填写对应的信息
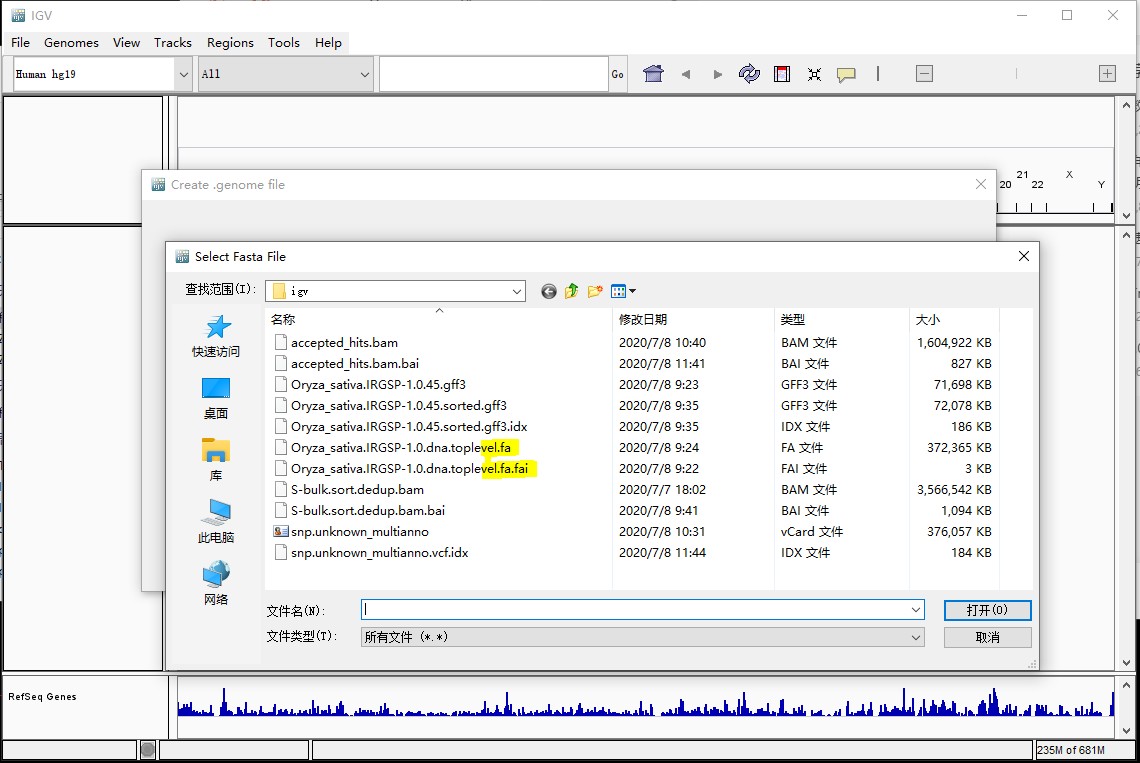
注意:原始的fasta文件要经过samtools工具处理建立一个后缀为.fai的文件(方法参考:https://www.omicsclass.com/article/1169)。并且这个.fai文件要和.fa文件在同一目录下。
添加完成后界面如图
三. 加载GFF注释文件
由于GFF文件很大,需要排序以及建立索引之后才能导入IGV:
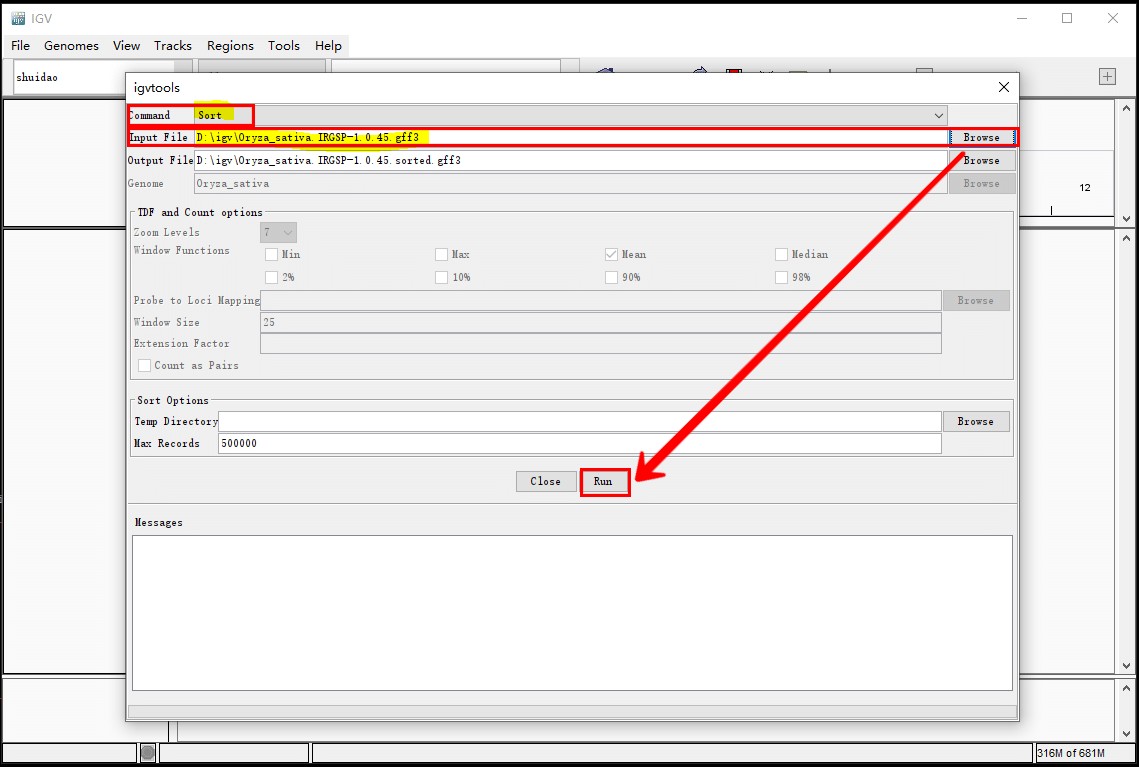
1.用igvtools对gff文件进行排序,得到这个文件
首先点击tools选项中的Run igvtools
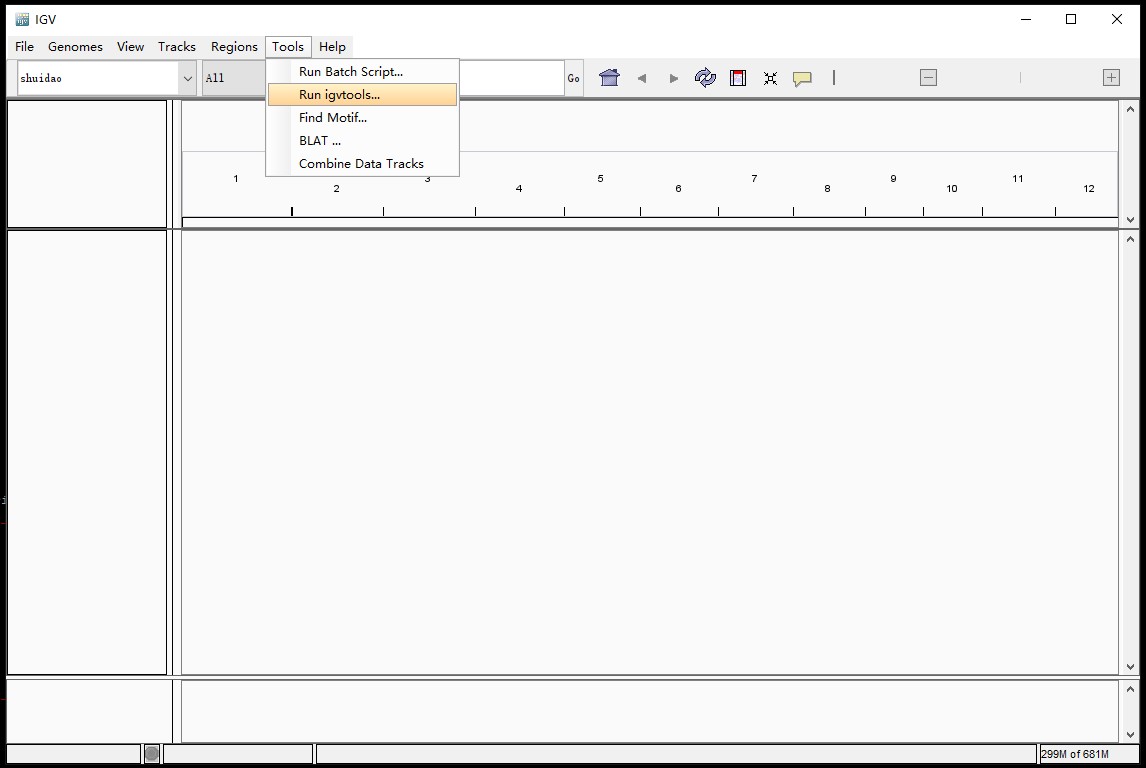
选择sort命令,输入文件为gff文件
然后对已排序的gff文件建立索引
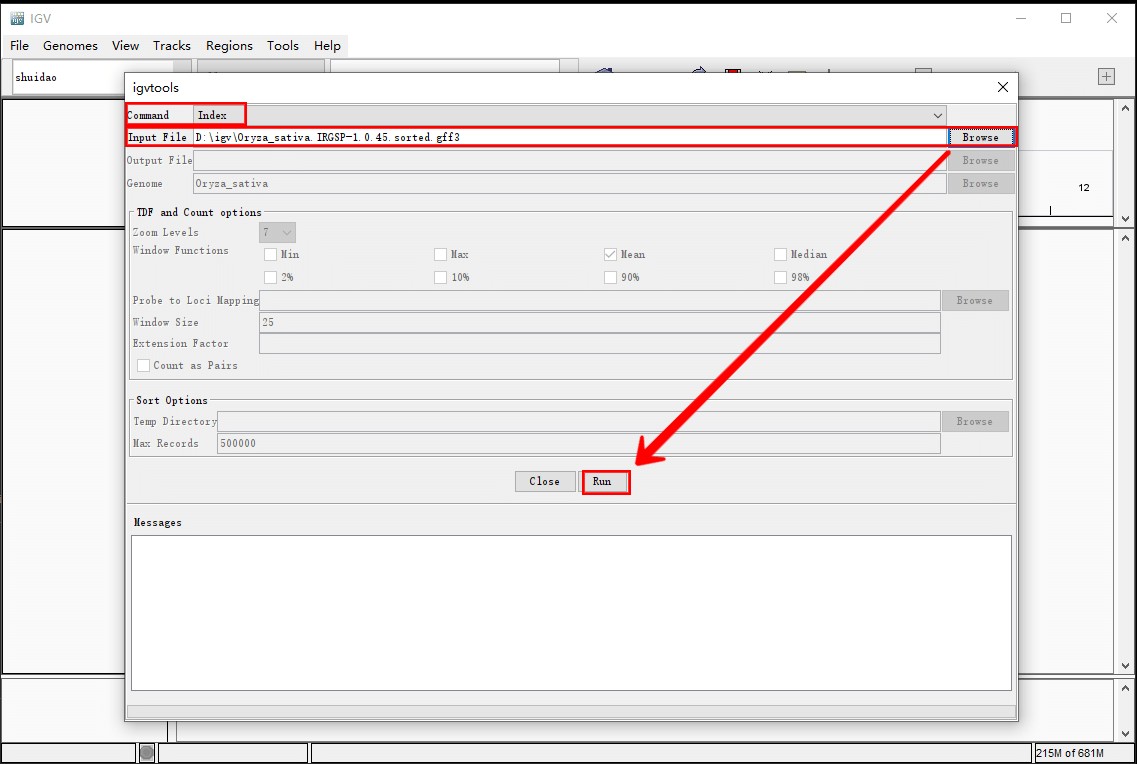
运行之后会得到一个.idx文件
最后点击File选项,选择第一个load from file添加排序的文件和索引的文件
添加之后,结果如图:
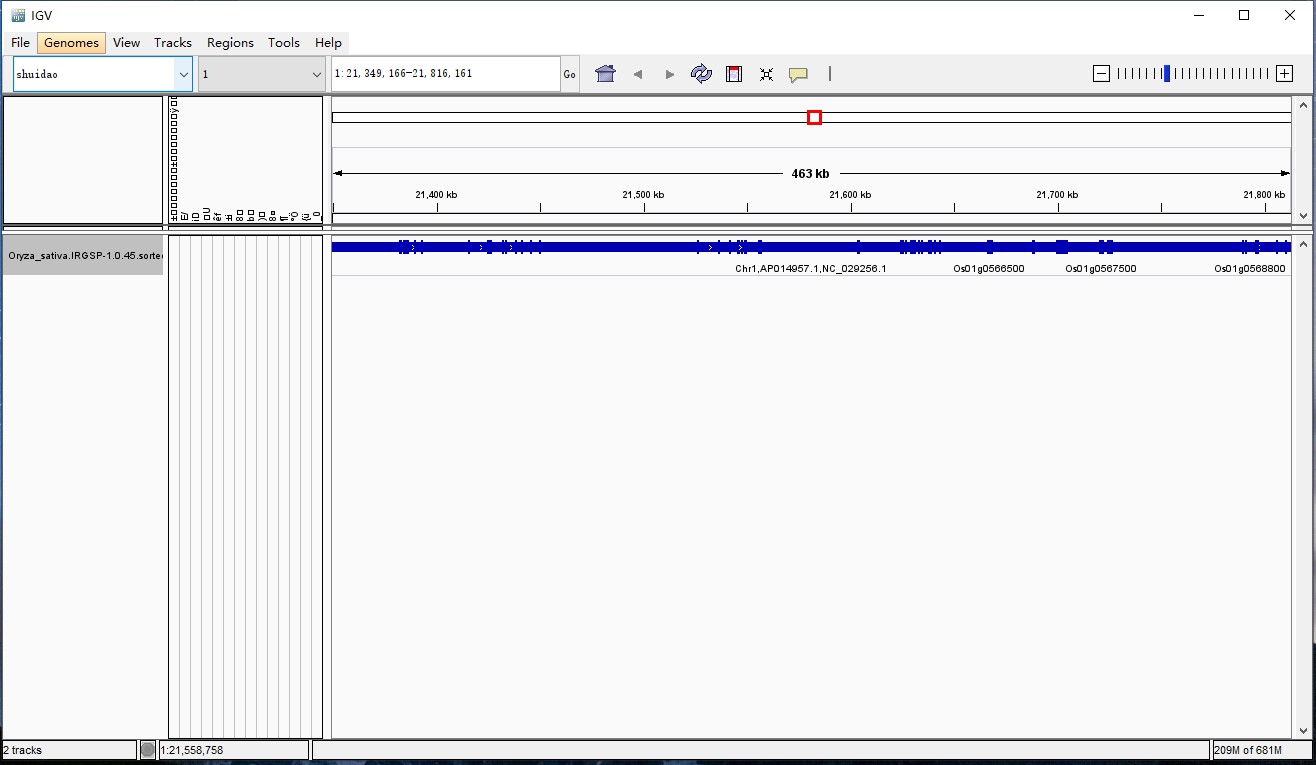
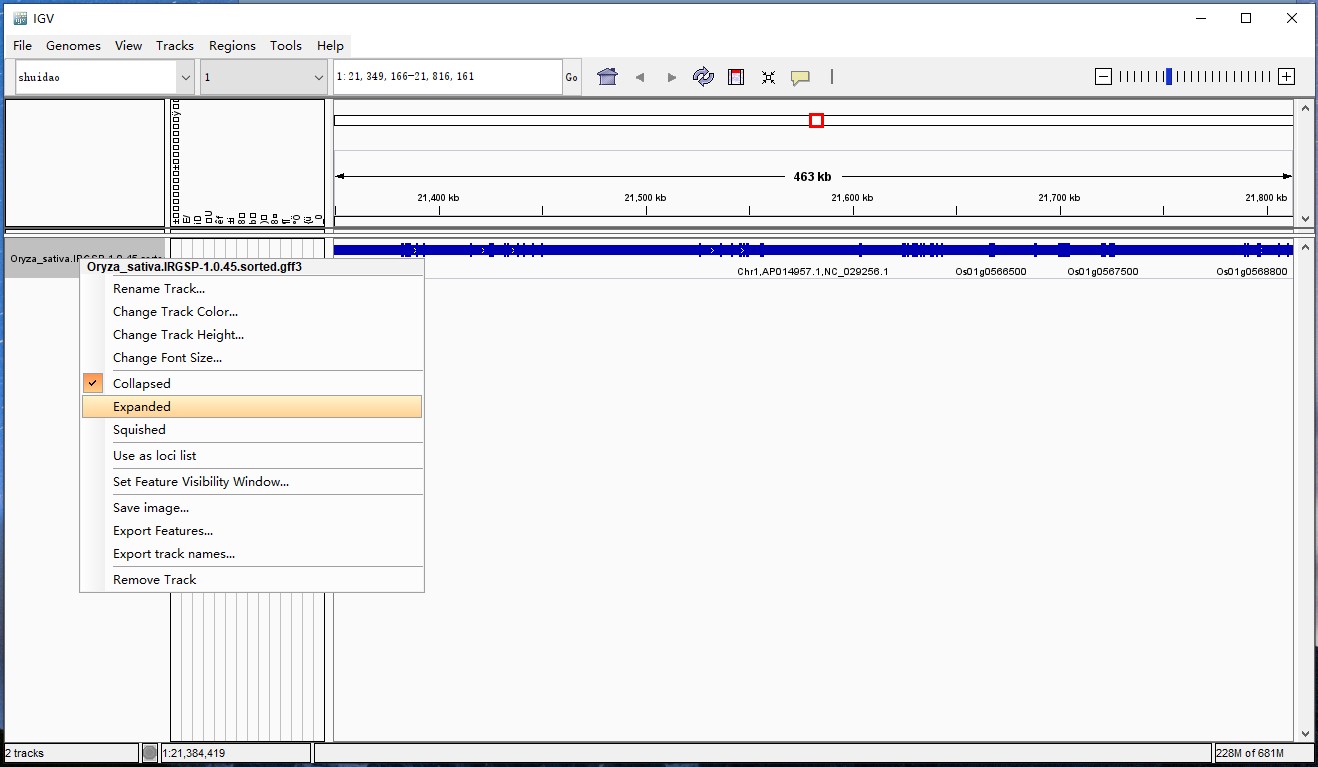
可右击track区选择Expanded更换详细显示模式
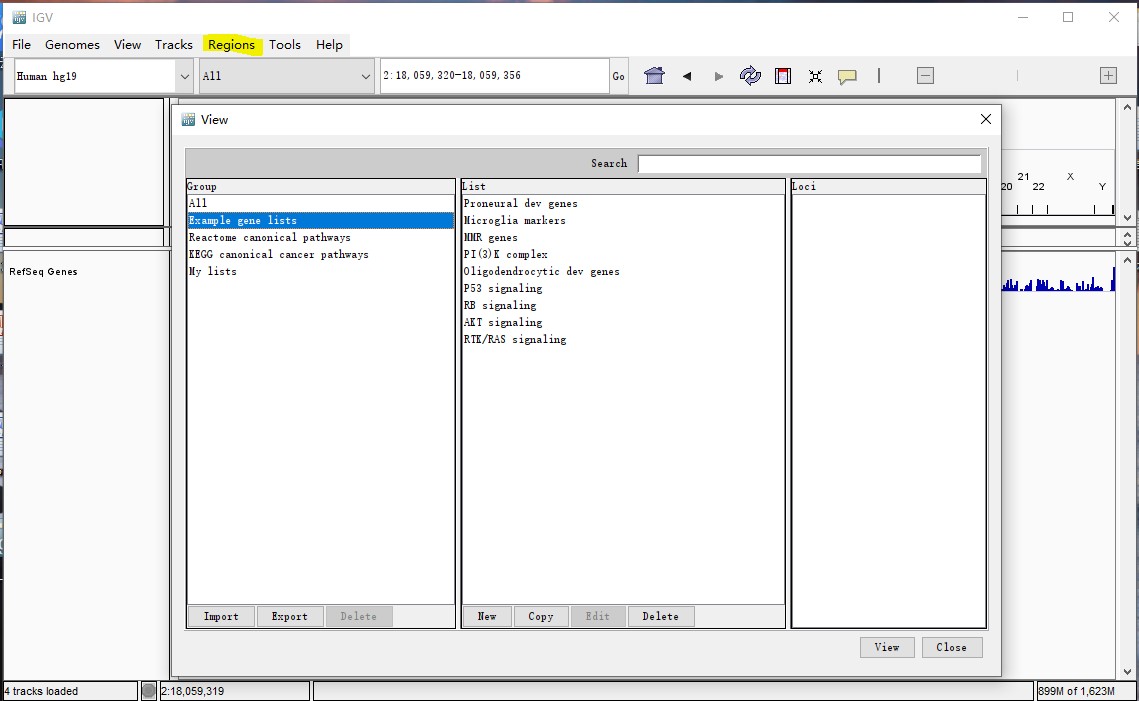
软件可选择基因集对指定基因进行可视化:点击Regions选项,选择Gene List,也可通过My lists 导入自己的基因集
四. 转录组比对bam文件查看
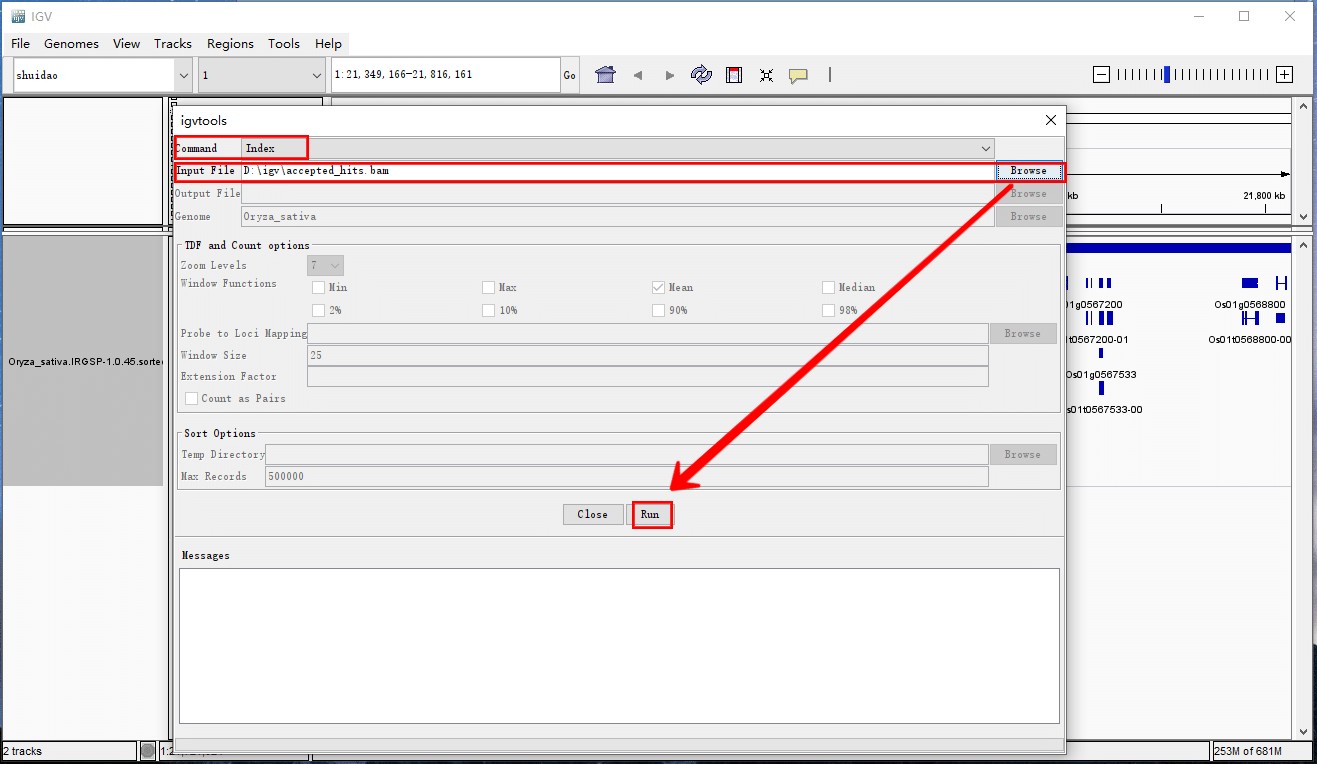
1.对bam文件构建索引:选择Tools选项,点击Run igvtools,选择index命令,选择bam文件
2.建立完索引后,点击File选项,选择第一个load from file添加bam文件,点击打开
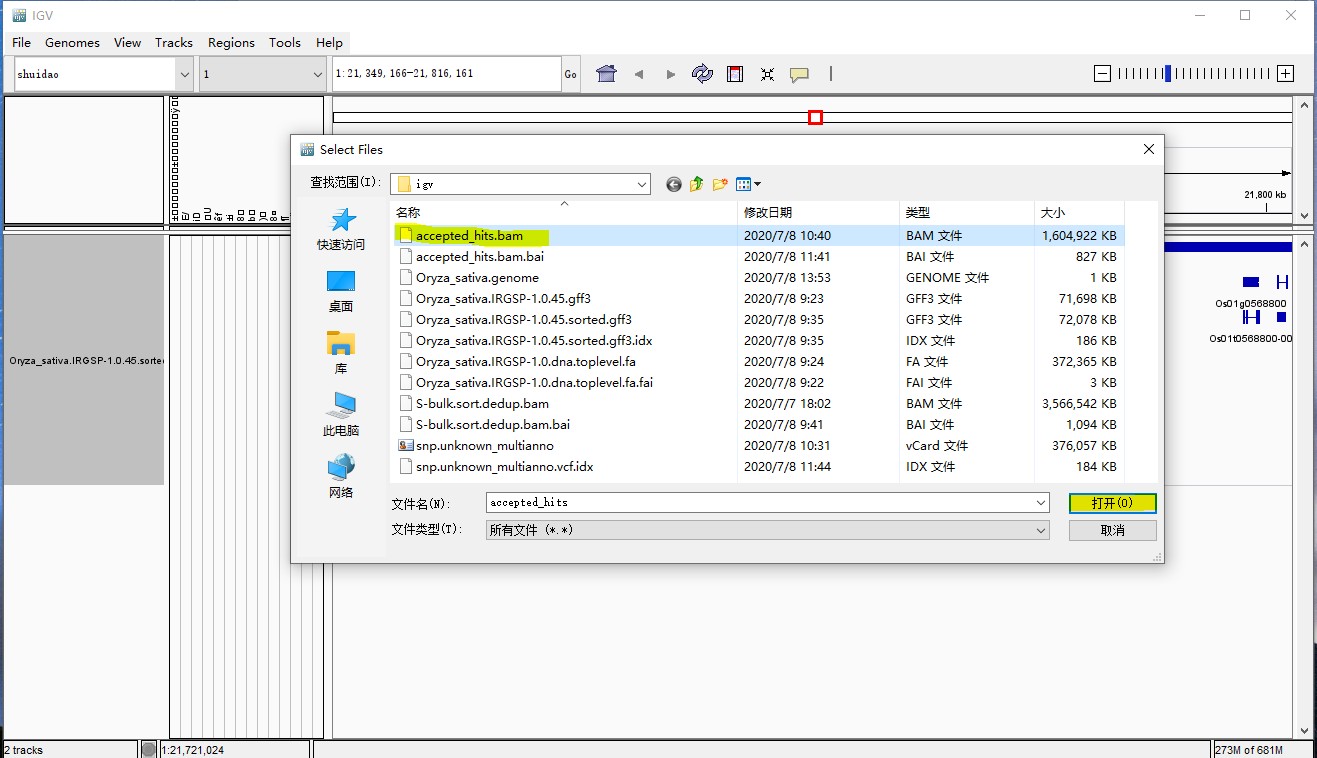
3.打开之后,可利用标尺显示位置,跳到指定的染色体位置,调节大小,查看具体信息。如果选择的区域过大,会显示zoom in to see coverage
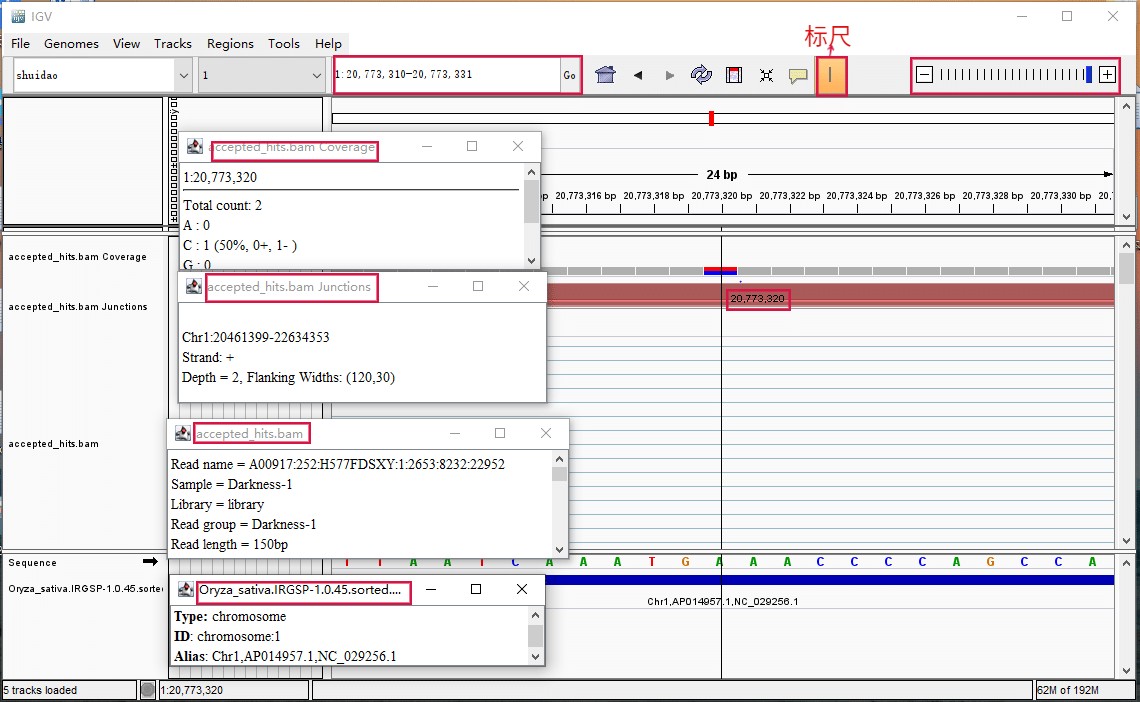
4.可点击 View选项下的 Preferences ,找到Alignments设置相关参数
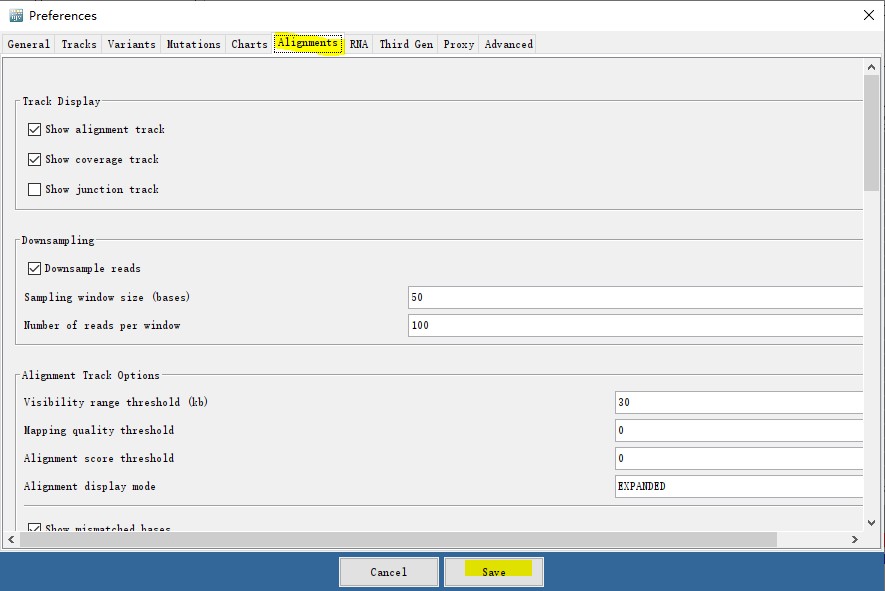
5.右击 Track 区进行 Color alignments by 的设置,在no color的情况下,alignments 只有亮灰和白色两种长条,其中白色的比对质量为零 ; 还可通过Sort alignments by 对Track区域进行排序(若要返回最初结果则选择 Re-pack alignments )以及Group alignments by对Track区域进行分组
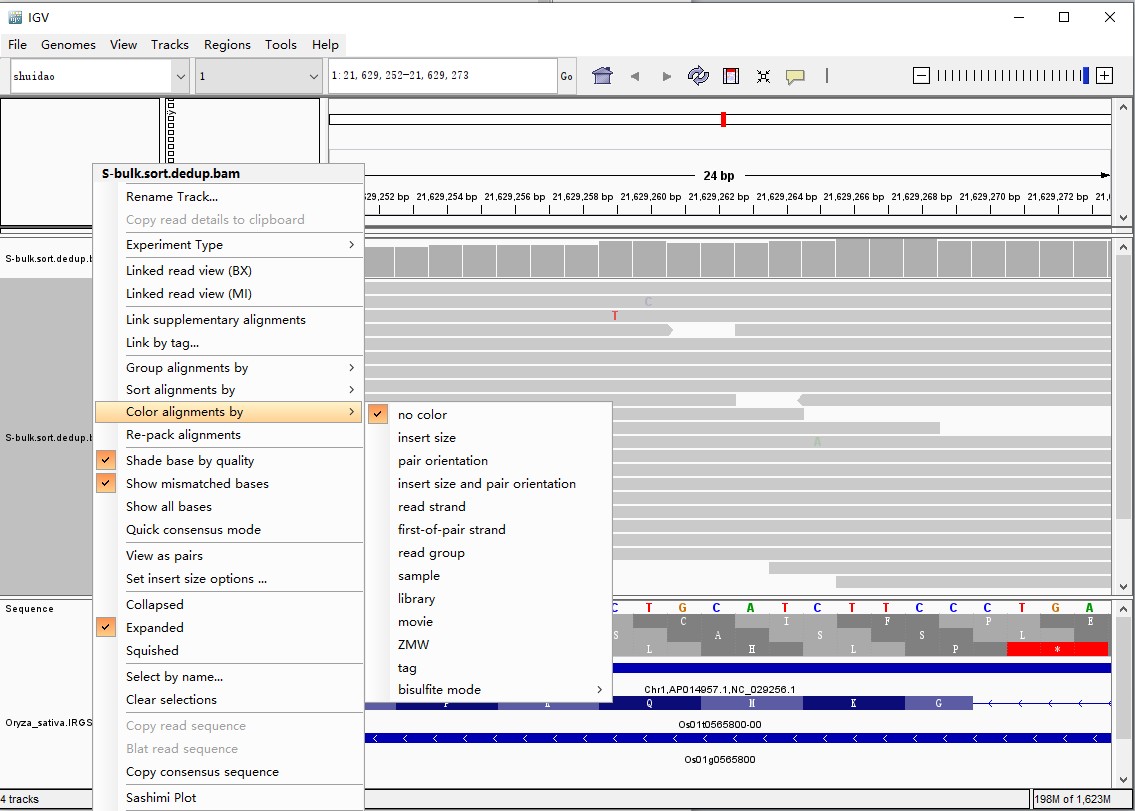
6. 如果发现存在与参考序列不同的碱基,会用不同的颜色标注出来,同一碱基颜色深浅表示碱基的质量,透明度越高说明检测的碱基质量越低,越不可信
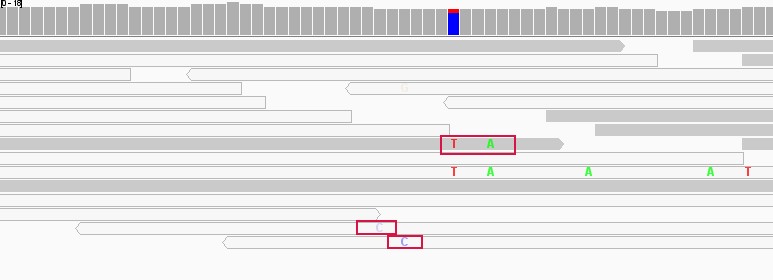
若发现存在与参考序列不同的碱基,软件会用不同的颜色标注出来,同一碱基颜色深浅表示碱基的质量,透明度越高说明检测的碱基质量越低,越不可信;插入片段用紫色的 I 或红色的 I (当插入的碱基数多余预设的阀值时)表示, 缺失用黑条表示

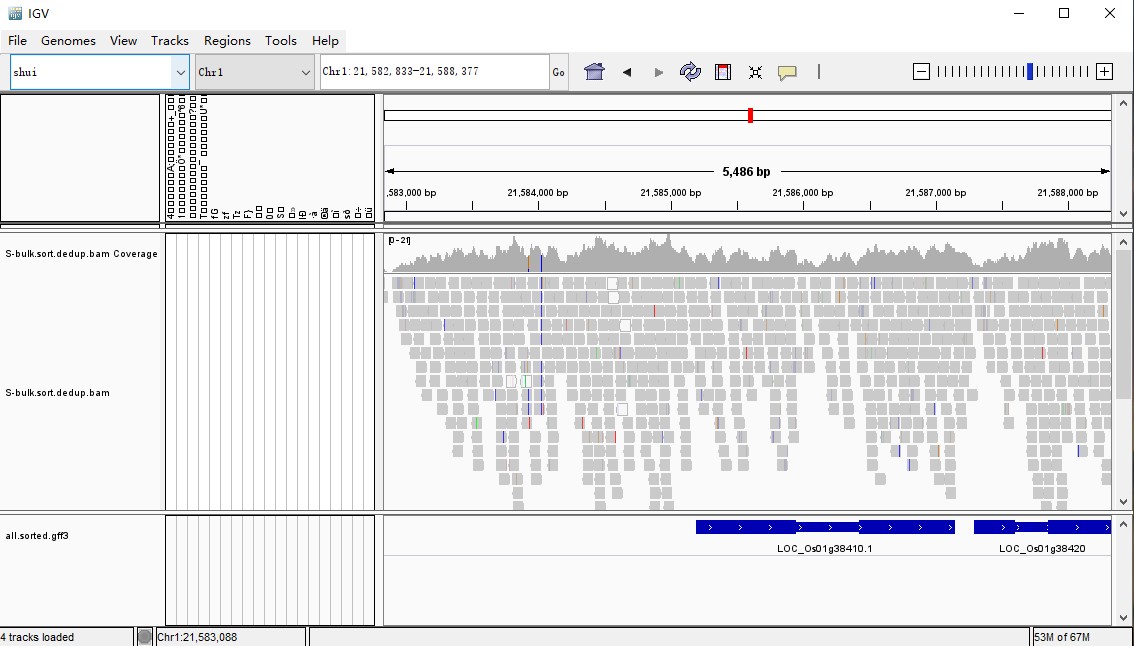
五. 重测序数据可视化
注意选择和测序数据一致的基因组,bam文件加载同上转录组方法,加载结果如下:
重测序变异结果查看:
1. 先用igvtools对vcf文件建立索引:选择Tools选项下的Run igvtools,选择index命令,选择vcf 文件建立索引。
2. 建立索引之后,点击File选项下的load from file 找到 vcf 文件打开,注意索引文件和vcf文件在同一目录下。
打开之后:
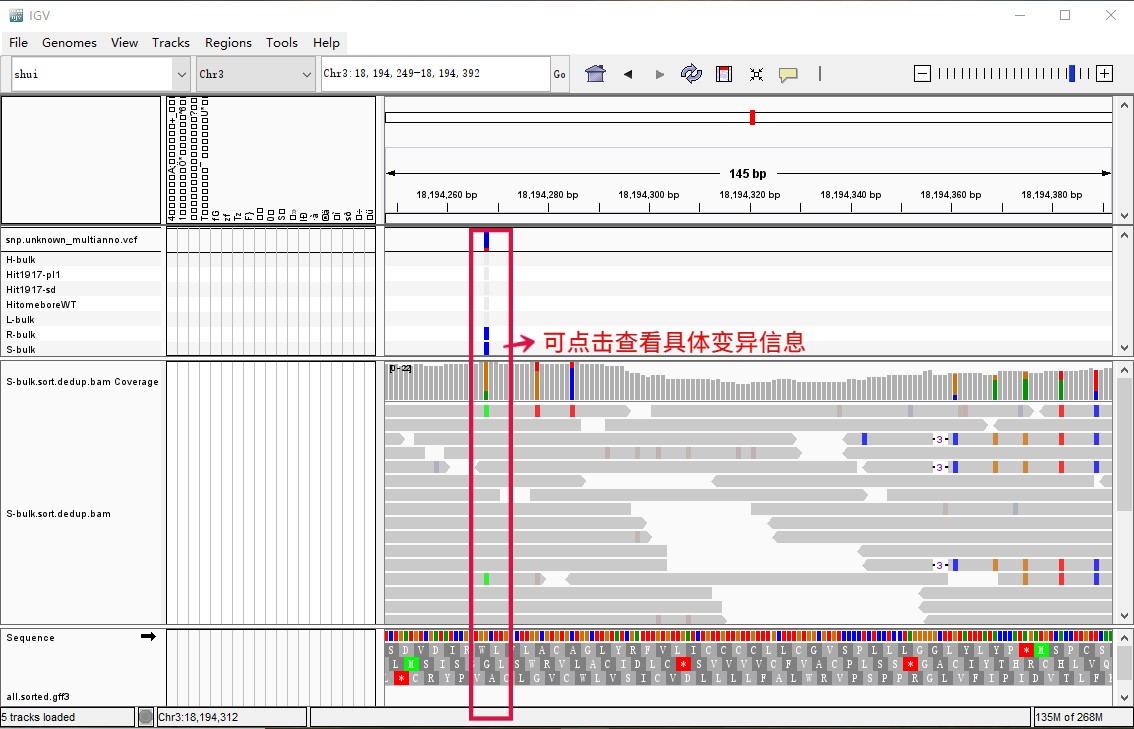
其他使用帮助:
http://www.broadinstitute.org/igv/book/export/html/6
http://www.broadinstitute.org/software/igv/UserGuide
更多生物信息分析课程:https://study.omicsclass.com/
- 发表于 2020-07-13 11:36
- 阅读 ( 17787 )
- 分类:软件工具
