GWAS中P值的筛选阈值原理是什么
GWAS关联分析课程推荐:https://bdtcd.xetslk.com/s/2KgXQq
对于统计学假设检验来说,多重检验校正是一个非常重要的领域。我们在进行假设检验的时候,通常会设置一个零假设,之后计算出一个p值,即数据分布符合原假设的概率,p值越低,即代表拒绝原假设的概率越大。我们通常认为p值<0.05是一个判断是否显著的阈值。但是在对多组数据同时进行比较的时候,简单地使用0.05作为阈值可能就不那么合适了。换句话说,在同时对多组数据进行处理和比较的时候,很可能其中部分数据因为随机效应而超过阈值,造成假阳性结果。而检验的次数越多,出现假阳性的概率就越大。而对于GWAS等对全基因组的位点进行统计学检验的分析,就急需一种方法来对结果的阈值进行校正。
Bonferroni校正即为最严格的多重检验矫正方法。其校正原理为:在同一数据集上同时检验n个相互独立的假设,那么用于每一假设的统计显著水平,应为仅检验一个假设时的显著水平的1/n。如以显著水平0.05检验同一数据集上两个独立的假设,此时用于检验该两个假设应使用更严格的0.025;对于10000个基因的检验,若将p设置为1e-6,进行10000次比较之后犯错误的概率是10-6*10000 = 0.01,严格地控制了假阳性的出现。
Bonferroni校正的主要应用范围就是GWAS,即全基因组关联分析。在全基因组关联分析中,如果对每个位点进行测试,以0.05的水平进行筛选,由上文可知,在1000000个位点中其假阳性的位点可能达到5%,即50000个,所以,为了控制假阳性的概率,以Bonferroni校正对阈值进行调整。Bonferroni校正会将设定的显著性水平除以测试次数,最终得到一个总的阈值。换句话说,它将阈值从a = 0.05调整为a =(0.05 / n),其中n是进行的统计检验的次数,即SNP的个数。最终得到的阈值根据进入分析的SNP数量不同会有较大的差异,但通常都会在1e-6以下的量级。
当然了,Bonferroni校正作为最严格的的多重假设检验校正方法,在实际应用当中也会出现一些问题。由于GWAS标记之间的连锁不平衡,可能会存在多个标记或者SNP之间相互连锁的情况,也就是说它们之间的分布并不是完全独立的,所以假设GWAS数据集的每个关联测试都是独立的是不正确的。因此,应用Bonferroni校正通常会为我们提供最保守的p值阈值。例如将1000000个位点纳入计算当中,Bonferroni校正所产生的阈值即为5e-8,由于其过为保守,往往会导致假阴性的产生,可能整个基因组中都很少有标记的关联p值能够达到这一标准。这时我们往往需要根据实际曼哈顿图的情况对阈值进行一些调整。
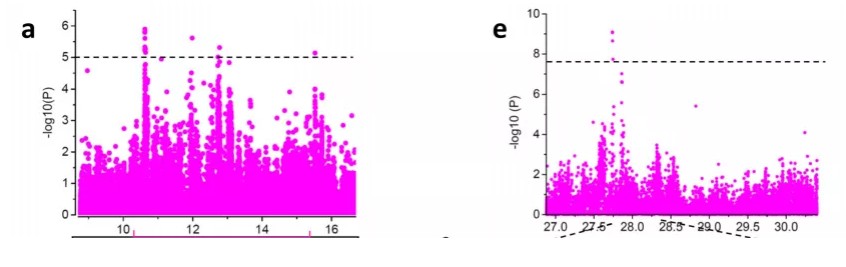
例如2019年Genome Biology桃重测序中的GWAS部分,大多数性状采用的均是经过Bonferroni校正的约3e-8的阈值,但是对于果实大小性状的GWAS分析,研究者将阈值降到了1e-5,以消除由于Bonferroni校正过于严格导致的假阴性。我们可以看到左图中虽然信号强度相对较低,但是有一簇SNP明显呈现连锁的信号,说明可能这里存在着一个与性状关联的QTL。
参考文献:
Li Y, Cao K, Zhu G, et al. Genomic analyses of an extensive collection of wild and cultivated accessions provide new insights into peach breeding history. Genome biology, 2019, 20(1): 36.
- 发表于 2020-11-12 13:37
- 阅读 ( 20595 )
- 分类:GWAS
