docker 工具高级用法
平时大家使用docker按照我们组学大讲堂课程中老师的操作:一般是启动相应的镜像并进入docker,我们的镜像默认进入/work目录,然后通过-v选项把我们系统里面的数据文件目录映射到容器中,这样就可以在/work目录分析数据了。但是仅仅会使用我们组学大讲堂提供的镜像有些局限,这里我给大家举例介绍一些大家在使用docker工具分析数据是的高级用法,方便大家更好的使用docker工具。
问题1:有时候我们运行一条命令可能需要很长时间,在这个过程中无意间关闭了终端,我的任务还在运行吗?还可以继续吗?
答:我们的任务还在运行,不必担心,docker容器还在后台保持运行。这时候我们还可以再进入那个容器,这里介绍操作命令:
> docker ps #查看后台正在运行的容器 CONTAINER ID IMAGE COMMAND CREATED STATUS PORTS NAMES a6c1ce4f6ae9 omicsclass/ampliseq-q2:v1.4 "/bin/bash" 24 seconds ago Up 23 seconds competent_payne > docker ps -a #-a 查看后台正在运行的或者挂掉的所有容器 我这里没有挂掉的容器所以只有一个 CONTAINER ID IMAGE COMMAND CREATED STATUS PORTS NAMES a6c1ce4f6ae9 omicsclass/ampliseq-q2:v1.4 "/bin/bash" 24 seconds ago Up 23 seconds competent_payne
这个时候可以使用 docker attach + 容器ID 再次进入那个容器,注意进入的状态和你关闭终端的状态一样。光标在闪烁,说明我关闭终端时候,那个任务还没有运行完。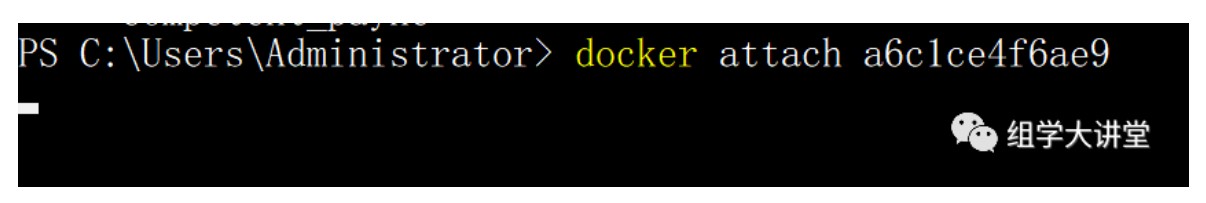
attach进入不是很好,这里介绍另一种进入正在运行的容器的方法:exec命令,意思是让容器执行一条命令,这里我们执行bash 即让容器新开一个bash终端;
> docker exec -it a6c1ce4f6ae9 bash
-it 为交互式,后面跟容器ID ,最后跟要执行的命令bash即可,此时我们进入容器相当于容器重新打开了一个终端。可以使用 ps xf 查看后台任务。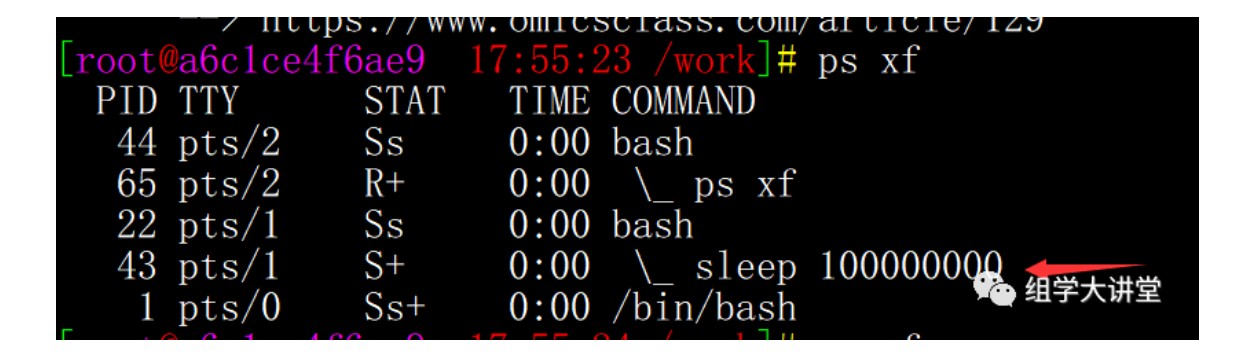
问题2:第三方Docker镜像不会使用,怎么办?
答:虽然我们组学大讲堂提供了很多镜像,安装了很多软件;但是,我们也不可能把所有人要求的软件全部安装,因此学会使用第三方提供的镜像也非常重要,免去自己安装软件的麻烦。如果使用第三方的镜像也就是其他人提供的镜像,那么绝大多数镜像都是最小安装(节约存储空间),也就是只安装了这个软件所需要的最小要求的软件环境,有的甚至连 wget less awk 等都没有安装,因此我们像我们课上讲的方法进入容器使用起来很不方便;所以使用这样的镜像,我们有另一种使用方法:不进入容器直接使用里面的命令就好了,例如:samtools 镜像,使用命令如下:
> docker search samtools #搜索第三方镜像 NAME DESCRIPTION STARS OFFICIAL AUTOMATED biocontainers/samtools Tools for manipulating next-generation seque… 11 [OK] mgibio/samtools-cwl Image containing samtools, for use in cwl wo… 3 [OK] pegi3s/samtools_bcftools SAMtools-BCFtools (http://www.htslib.org/) d… 2 kfdrc/samtools alpine based samtools 2 [OK] jweinstk/samtools Minimal docker container for samtools. 1 > docker pull biocontainers/samtools #下载镜像 > docker images #查看后台镜像 已经下载完成 REPOSITORY TAG IMAGE ID CREATED SIZE omicsclass/ampliseq-q2 v1.4 d18a28c0f559 4 weeks ago 6.7GB omicsclass/biocontainer-base latest d7659e1b1b1f 3 months ago 1.83GB omicsclass/ampliseq-q1 v1.2 e2969668cba3 4 months ago 10.4GB biocontainers/samtools latest a44635d66fda 2 years ago 1.21GB
以下为关键操作步骤:找个fasta文件放在D:\samtools目录下,如下图所示,我们的任务是使用samtools对这个文件建立索引:
docker使用命令如下:
> docker run --rm -v D:/samtools/:/work -w /work biocontainers/samtools:latest samtools faidx dna-sequences.fasta
注意:-w 选项 指定容器中的进入的工作路径,把我们映射的路径指定为工作路径(/work)。然后把所有的输入文件都放在D:\samtools目录下,这时候我们的容器里面的/work 目录即是D:\samtools。
这样就像运行本地命令一样运行samtools,只是前面多了一串docker的命令,查看结果:
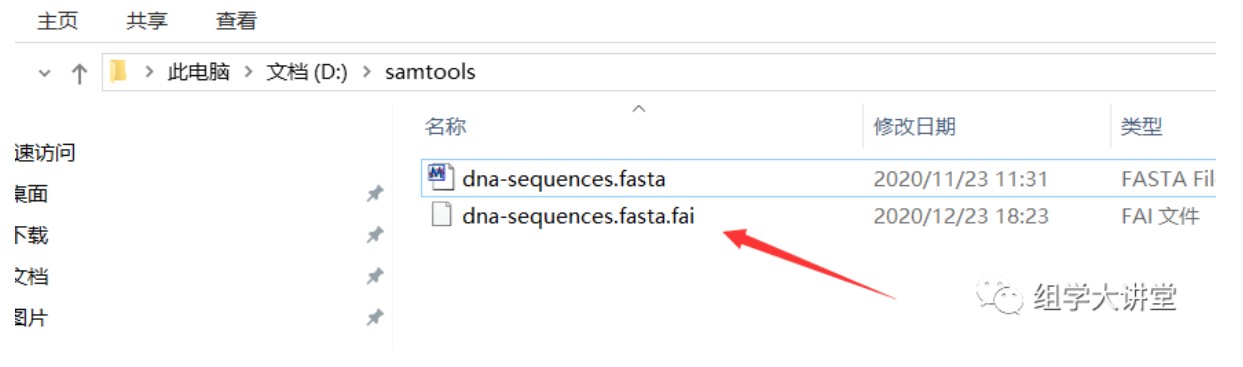
问题3:有很多命令而且运行时间很长,怎么快速运行?
答:当然可以的,我们可以把我们要用全部的命令都写到一个sh文件里面,让docker容器在后台批量运行即可;我们组学大讲堂提供的实操课程中的数据分析都可以这样后台批量运行work.sh,下面举例:
#-d 参数表示后台运行容器,-it 表示交互式进入容器这里没有设置 > docker run --rm -d -m 6G --cpus 2 -v D:/qiime1-16s:/work -w /work/amplicon_demo omicsclass/ampliseq-q1:v1.2 sh work.sh 53be3e69d2d6f6e0b2fed2e7a9e528a8465f653bd294809dcbe017427bca3bcd > docker ps #查看后台运行的镜像 CONTAINER ID IMAGE COMMAND CREATED STATUS PORTS NAMES 53be3e69d2d6 omicsclass/ampliseq-q2:v1.4 "sh work.sh" 2 seconds ago Up 2 seconds nice_curran > docker logs 53be3e69d2d6 #查看运行日志 mkdir: cannot create directory ‘/work/amplicon_demo/tmp’: File exists mkdir: cannot create directory ‘/work/amplicon_demo/1.import_data’: File exists Imported /work/amplicon_demo/manifest.ckd.txt as PairedEndFastqManifestPhred33V2 to demux.qza > docker rm -f 53be3e69d2d6 #终止运行容器,可直接删除
docker 安装使用视频课程
安装过程可观看视频课程操作演示:

- 发表于 2020-12-24 17:36
- 阅读 ( 8635 )
- 分类:linux
