blast比对软件使用
一、下载
下载地址:ftp://ftp.ncbi.nlm.nih.gov/blast/executables/blast+/LATEST
选择需要的版本进行安装即可
二、使用
1. 建库
makeblastdb -in genome.cds.fa -dbtype nucl -title genome.cds.fa #DNA序列makeblastdb -in genome.pep.fa -dbtype prot -title genome.pep.fa #蛋白质序列
说明:
-in:建库序列
-dbtype:数据库类型
以蛋白质序列为例,运行结果:
2 . 比对
blastall -i Aphidoidea.fa -d genome.cds.fa -p blastn -e 1e-10 -b 5 -v 5 -m 8 -o yachong.blast
说明:
-i:需要比对的文件的路径
-d:数据库的路径
-e:期望值,这一参数控制搜索的灵敏度
-b:设定输出文件中,最多显示多少个query-subject两两比对文本描述,默认值 为250
-v:设定输出文件中,匹配列表最多显示多少个subject,默认值为500
-m:设定搜索结果的显示格式,m参数的选项有12个(通常用-m8格式)
-o:输出文件的路径
-p : 可选择blast的方法:blastp,blastx,blastn
| -p | query | database | |
| blastn | 核酸 | 核酸 | |
| blastp | 蛋白质 | 蛋白质 | |
| blastx |
| 蛋白质 | |
| tblastn | 蛋白质 |
|
3.比对结果:
运行代码之后,会生成文件 yachong.blast,文件内容如下:
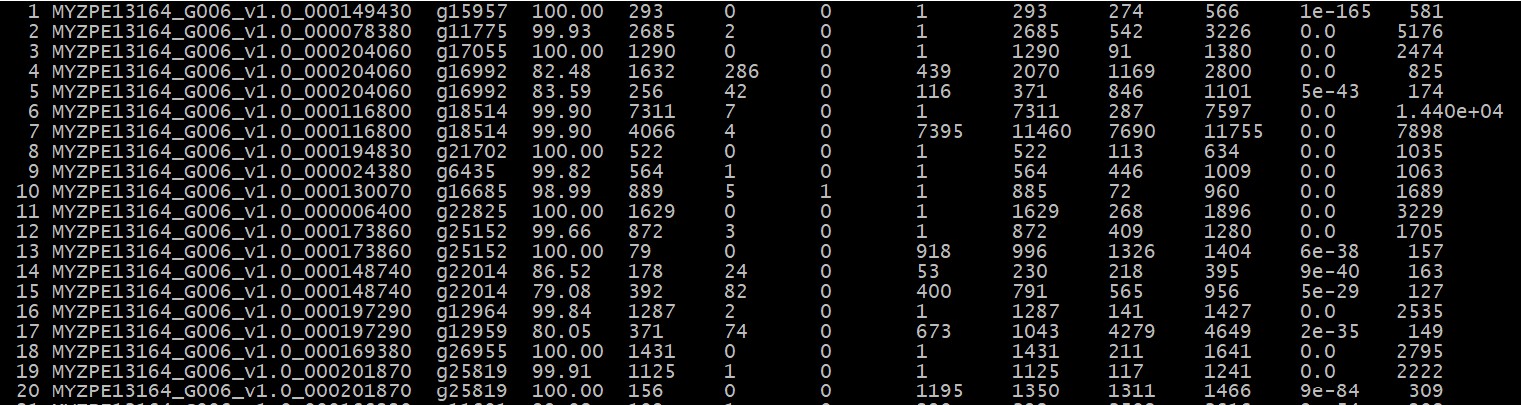
一共有12 列,分别代表:
1、Query id:查询序列ID标识
2、Subject id:比对上的目标序列ID标识
3、% identity:序列比对的一致性百分比
4、alignment length:符合比对的比对区域的长度
5、mismatches:比对区域的错配数
6、gap openings:比对区域的gap数目
7、q. start:比对区域在查询序列(Query id)上的起始位点
8、q. end:比对区域在查询序列(Query id)上的终止位点
9、s. start:比对区域在目标序列(Subject id)上的起始位点
10、s. end:比对区域在目标序列(Subject id)上的终止位点
11、e-value:比对结果的期望值
12、bit score:比对结果的bit score值
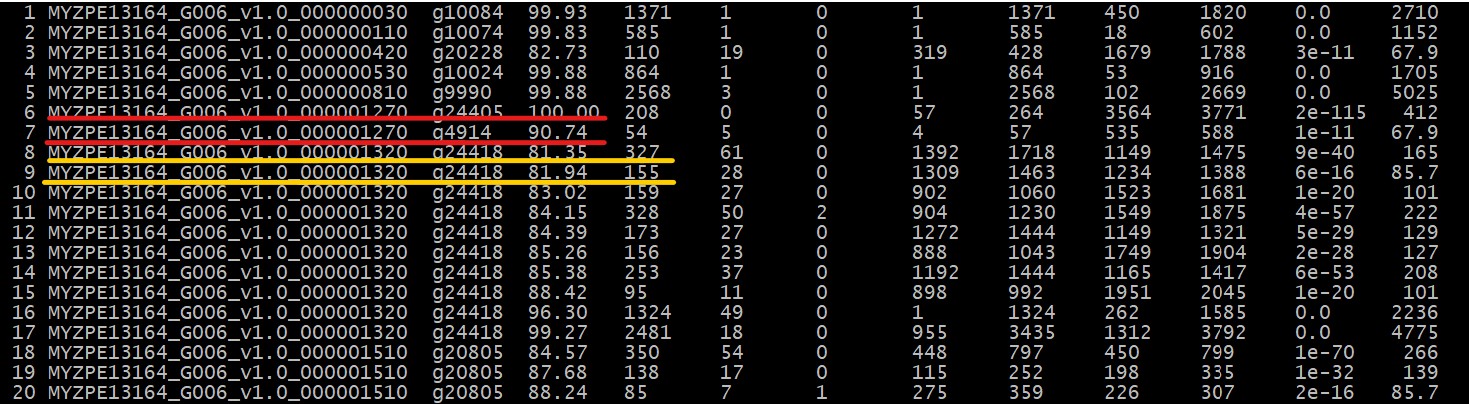
注意:结果会出现一条查询序列与多条目标序列相似的情况,挑选打分高即可(红色线条部分);还会出现一条查询序列与同一条目标序列不同位置相似的情况(黄色线条部分)。
此外:
blast还有在线版:https://blast.ncbi.nlm.nih.gov/Blast.cgi
使用参考:https://www.omicsclass.com/article/1192
此外,我们在网易云课堂上有各种教学视频,有兴趣可以了解一下:
1. 文章越来越难发?是你没发现新思路,基因家族分析发2-4分文章简单快速,学习链接:基因家族分析实操课程
2. 转录组数据理解不深入?图表看不懂?点击链接学习深入解读数据结果文件,学习链接:转录组(有参)结果解读;转录组(无参)结果解读
3. 转录组数据深入挖掘技能-WGCNA,提升你的文章档次,学习链接:WGCNA-加权基因共表达网络分析
4. 转录组数据怎么挖掘?学习链接:转录组标准分析后的数据挖掘
6. 更多学习内容:linux、perl、R语言画图,更多免费课程请点击以下链接:
http://m.study.163.com/provider/400000000234009/index.htm?share=1&shareId=1433306807
- 发表于 2021-02-22 14:29
- 阅读 ( 4733 )
- 分类:软件工具
