蛋白3D结构预测 3D Protein Structure Prediction
- 初级结构——氨基酸序列;
- 二级结构——а螺旋(alpha-helix),β折叠(β-sheets),β转角,无规则卷曲(random coil)
- 三级结构——三维结构,由模体(motif)和结构域(domain)组成;
- 四级结构——亚基之间的互作。
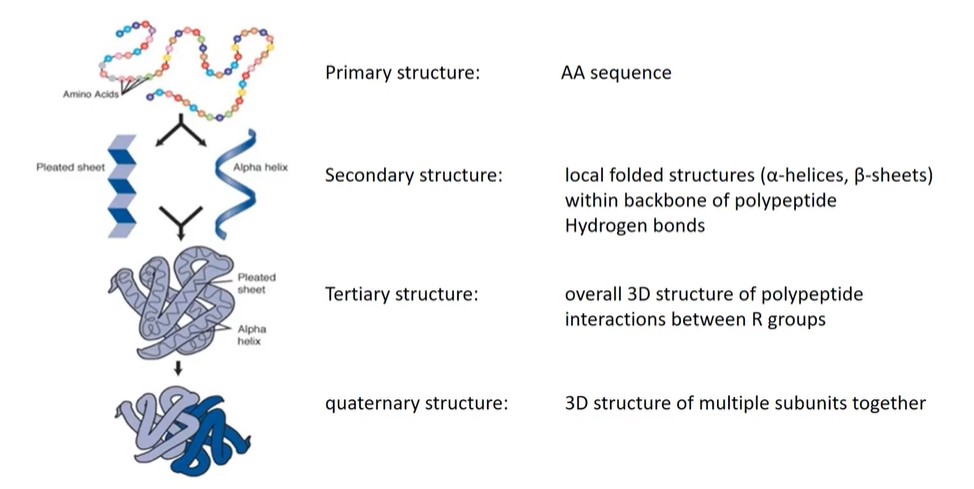
蛋白质的生物活性不仅决定于蛋白质分子的一级结构,而且与其特定的空间结构密切相关。异常的蛋白质空间结构很可能导致其生物活性的降低、丧失,甚至会导致疾病,疯牛病,Alzheimer's症等都是由于蛋白质折叠异常引起的疾病。强调活体细胞内的蛋白质正常折叠、异常折叠的研究,尤其是折叠催化剂、分子伴侣和大分子的参与是这一领域研究热点。在功能和结构细节上阐明关于蛋白质折叠的过程将对相关疾病的预防和治疗有重要意义。
一 蛋白质三级结构预测方法:
1.同源模建 homology modeling :
相似的氨基酸序列对应着相似的蛋白质结构 ,找到与目标序列一致度≥30%已知结构作为模板代表工具: SWISS-MODEL https://swissmodel.expasy.org/
其他工具:

同源蛋白模型构建(模建)的步骤:
① 目标蛋白序列与目标序列的匹配:应用 FASTA 或 BLAST 搜索软件,在 PIR 、 SWISSPROT 或 GENEBANK 等序列库中按序列同源性挑选出一些同源性比较高的序列,然后把挑选出的序列与目标序列基序多重匹配,得到模板结构等价位点套的初始集合。
② 根据模板结构构建目标蛋白结构模型:在已确定的模板结构等价位点套的初始集合的基础上,旋转每一个模板的结构,使它们相互间的位置尽可能多地重叠在一起。不同两个模板在空间中若复合一定的重叠距离标准,那它们相互之间的关系就是等价位点。许多这样的等价位点构成了等价位点套。
叠合结束后,即得到了同源蛋白的结构保守区( SCRs ),以及相应的基架结构( framework )。模板结构匹配后,一般还要用得到的同源体的 SCRs 的第一条序列与目标序列匹配,挑选出目标序列上的高相拟区,定义为目标蛋白的 SCRs 。Homology 、 UQANTA/CHARM 、 COMPOSER 、 CONSENSUS 、 MODELLER 和 Collar extension 等软件和方法可以用于目标蛋白结构模型的构建。
③ 对模建结构基序优化和评估:同源结构模建(预测)得到的蛋白质结构模型,通常含有一些不合理的原子间接触,需要对模型进行分子力学和分子动力学优化,消除模型中不合理的接触。另外,模型中有些键长、键角和二面角也有可能不合理,也需要检查评估。PROCHECK 和 PROSA II 等软件常用于完成这类工作。
2. 折叠识别 fold recognition (穿线法 threading):
原理:不相似的氨基酸序列也可以对应着相似的蛋白质结构。已知的蛋白质结构有十几万个,但其所具有的不同的结构拓扑只有1393个,也就是说,所有结构都落在这1393个拓扑内!因此,选择匹配能量最低的拓扑。一般是不能同源建模(一致度<30%)的蛋白选用这个方法。
代表工具: I-TASSER (Iterative Threading ASSEmbly Refinement) https://zhanglab.ccmb.med.umich.edu/I-TASSER/
折叠法是近年来发展起来的一种比较新的方法。它可以应用到没有同源结构的情况中,且不需要预测二级结构,即直接预测三级结构,从而可以绕过现阶段二级结构预测准确性不超过 65% 的限度,是一种有潜力的预测方法。
反向折叠法的主要原理是把未知蛋白的序列和已知的结构进行匹配,找出一种或几种匹配最好的结构作为未知蛋白质的预测结构。它的实现过程是总结出已知的独立的蛋白质结构模式做为未知结构进行匹配的模板,然后用经过对现有的数据库的学习总结出的可以区分正误结构的平均势函数( Mean Force Field ),做为判别标准来选择出最佳的匹配方式。这种方法的局限性在于它假设蛋白质折叠类型是有限的,所以只有未知蛋白质和已知蛋白质结构相像的时候,才有可能预测出未知的蛋白质结构。如未知蛋白质结构是现在还没有出现的结构类型时,这种方法将不能被应用。
3. 从头计算 ab initio method
原理:1973年《science》Anfinsen:蛋白质的三维结构决定于自身的氨基酸序列,并且处于最低自由能状态。模拟肽段在三维空间中所有可能的姿态,并计算出自由能最低的一个。计算量极大,不常用。代表工具QUARK https://zhanglab.ccmb.med.umich.edu/QUARK/
二 以SWISS-MODEL 网站构建蛋白3D模型为例
1.方法步骤: 输入数据
2.同源模板选择
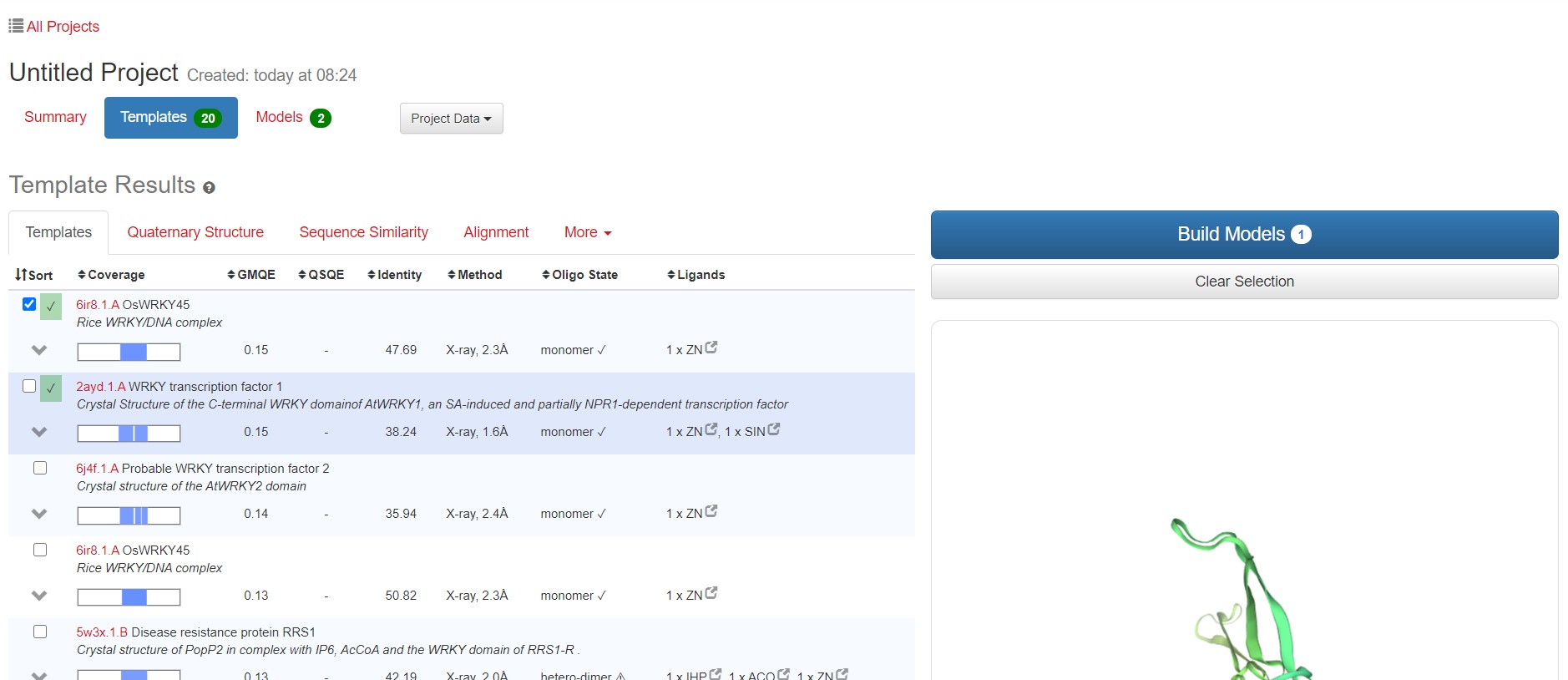
GMQE(全局模型质量估计)是一种结合目标-模板对齐方式和模板搜索方法的属性的质量估计。所得的GMQE分数表示为0到1之间的数字,反映了使用该对齐方式和模板构建的模型的预期准确性以及目标的覆盖范围。数字越高表示可靠性越高。可信度范围为 0-1,值越大表明质量越好。
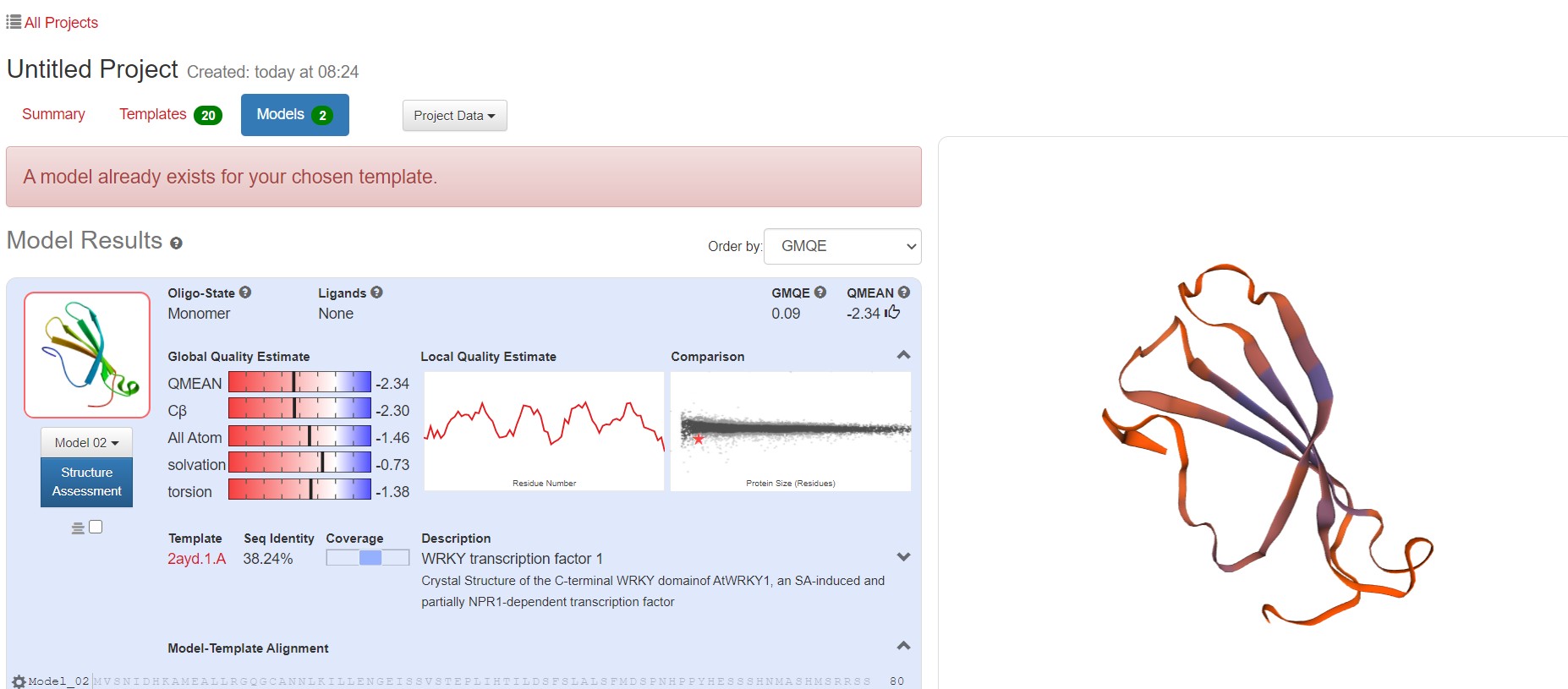
3.根据标准选择结果最好的model
点开某个模型,指标有很多,这里最重要的是:QMEAN。QMEAN的得分可与相似大小的实验结构所期望的得分相媲美。0值附近的QMEAN 得分表明模型结构与相似大小的实验结构之间具有良好的一致性。分数为-4.0或以下表示模型的质量较低。区间-4-0,越接近0,评估待测蛋白与模板蛋白的匹配度越好。
4.结果保存
选择好合适的模型即可构建3D模型,之后保存模型的pdb文件:
图片保存
三.第三方软件对模型评分
模型预测出来后需要有评估软件认为合格才能用,下载PDB文件,提交到测评软件。
SAVES:(一次性提供6个软件评估结果)https://saves.mbi.ucla.edu/ ,其中有三个显示通过即表示模型可用。多个软件判断模型好坏依据。
1).verify 3D
超过80%的残基拥有大于0.2的3D/1D值,则模型质量合格,低于0.2的部分需要进一步修正。
2).procheck
PROCHECK程序不考虑能量,只检测结构中的残基之间角度是否合理,生成Ramachandran plot。PROCHECK显示氨基酸残基核心区:91.3%,允许区:8.3%,大致允许区:0.4%,禁阻区:0%,位于可接受区的达到了100%,一般位于可接受区的氨基酸残基大于90%可以认为蛋白结构合理。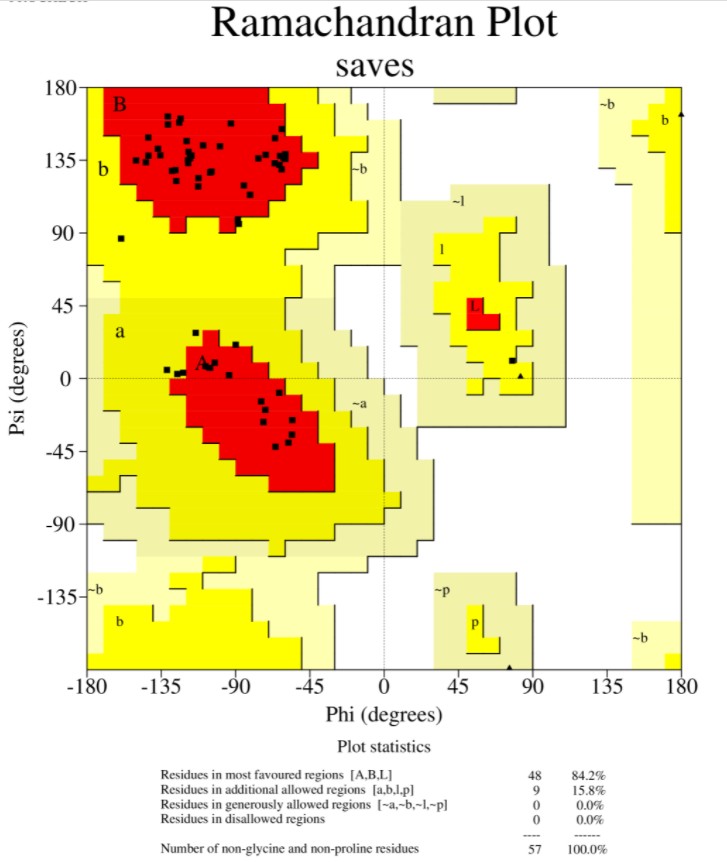
3).whatcheck
提交的蛋白结构与正常结构之间的差异,指标贼多,绿色多就当通过了。
4).errat
计算0.35nm范围之内,不同的原子类型对之间形成的非键相互作用的数目(侧链)。得分>85较好,晶体可达到95,一般来说结果在91以内。
5).prove
与预先计算好的一系列标准体积的差别,用z-score来表示,显示模版蛋白质与待测蛋白之间的匹配程度,越高越好。
四 常用蛋白序列和结构数据库网址链接 :
PDB 蛋白质三维结构 http://www.rcsb.org/pdb
SWISS-PROT 蛋白质序列数据库 http://kr.expasy.org/spro t/
PIR 蛋白质序列数据库 http://pir.georgetown.edu/
OWL 非冗余蛋白质序列 http://www.bioinf.man.ac.uk/dbbrowser/OWL/
EMBL 核酸序列数据库 http://www.embl-heidelberg.de/
TrEMBLEMBL 的翻译数据库 http://kr.expasy.org/sprot/
GenBANK 核酸序列数据库 http://www.ncbi.nih.gov/Genbank/
PROSITE 蛋白质功能位点 http://kr.expasy.org/prosite/
SWISS-MODEL 从序列模建结构 http://www.expasy.org/swissmod/SWISS-MODEL.html
SWISS-3DIMAGE 三维结构图示 http://us.expasy.org/sw3d/
DSSP 蛋白质二级结构参数 http://www.cmbi.kun.nl/gv/dssp/
FSSP 已知空间结构的蛋白质家族 http://www.ebi.ac.uk/dali/fssp/fssp.html
SCOP 蛋白质分类数据库 http://scop.mrc-lmb.cam.ac.uk/scop/
CATH 蛋白质分类数据库 http://www.biochem.ucl.ac.uk/bsm/cath/
Pfam 蛋白质家族和结构域 http://pfam.wustl.edu/蛋白质数据库( HPDB ) http://hpdb.hbu.edu.cn/
- 发表于 2021-04-12 13:36
- 阅读 ( 27854 )
- 分类:基因家族分析
