转录组扫盲系列--转录组如何建库?
很多老师都做过转录组,但你对转录组了解吗?知其然,更要知其所以然,才能对数据结果理解更深入,从本期起,小编给大家详细介绍下转录组实验及分析过程,里面肯定有你不知道的知识,赶紧get下吧!今天先介绍转录组建库原理。
RNA提取
转录组测得mRNA,提取却是提取的总RNA,只有质量合格的RNA,才会用来建库,评判RNA质量的有总量、浓度、有无杂质、RNA完整度,RNA质量标准参见推文:什么是RIN值?
真核生物体中总RNA=(~90%)rRNA+ (1~2%)mRNA+(8~9%)其他RNA,对于真核生物,成熟的mRNA可通过 oligo dT磁珠富集mRNA来进行建库;而如果想研究lncRNA、全转录组或原核生物转录组,可以通过去除rRNA的方法进行建库。
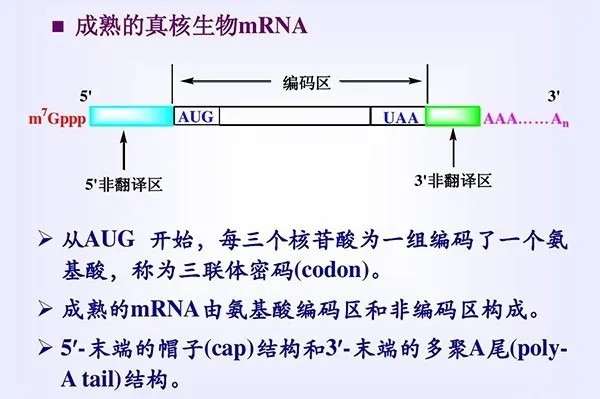
真核转录组建库
经常有人问我建库是先反转录还是先打断,今天就给大家一个明确的答案,真核转录组建库是先打断,然后再反转录,建库流程如下:
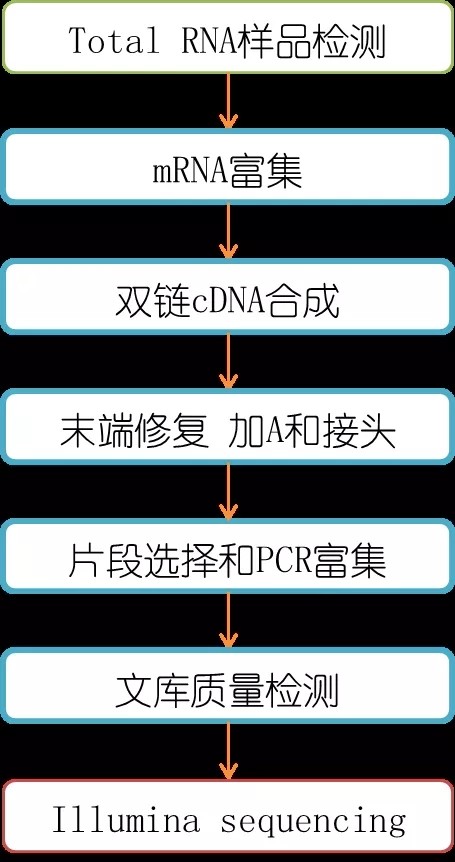
1. mRNA富集:RNA检测合格后,首先用带有Oligo(dT)的磁珠,通过A-T互补配对与mRNA的ployA尾结合的方式富集真核生物的mRNA;
2. 片段化:加入fragmentation buffer试剂将mRNA打断成短片段;
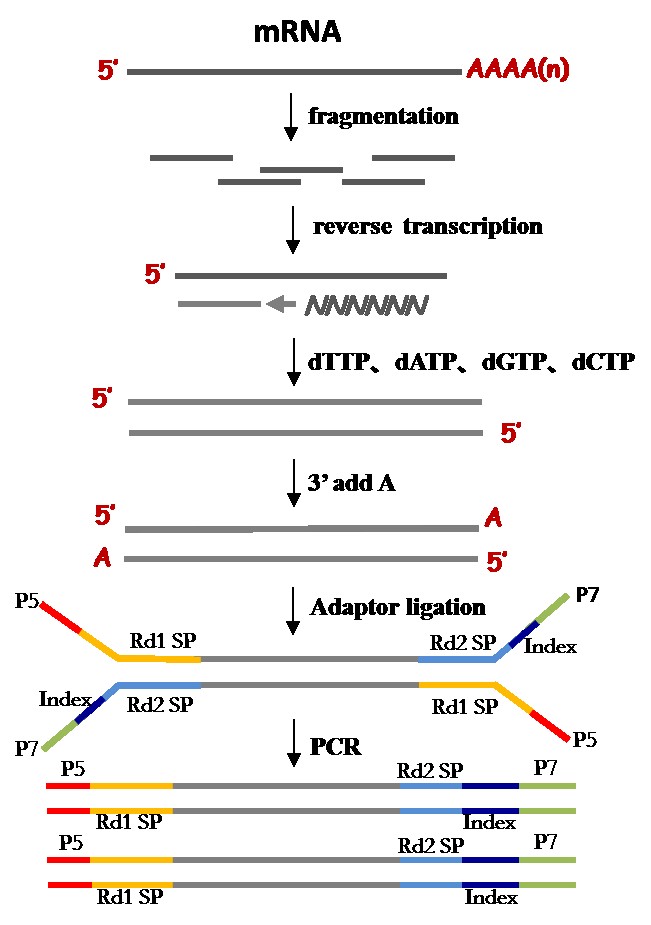
3. 反转录:以mRNA为模板,用六碱基随机引物(random hexamers)合成一链cDNA,然后加入缓冲液、dNTPs和DNA polymerase I合成二链cDNA,随后利用AMPure XP beads纯化双链cDNA;
4. 末端修复:末端补齐,再纯化修复后的cDNA,cDNA3‘端加A(dA - tailing);
5. 连接接头:每个接头都有一个6bp的index,也叫barcode,每个样品用不同index接头,这样即使将不同样品混在一同测序(混之前要归一化),也可以再根据index区分出来。
6. 片段选择和PCR富集:用AMPure XP beads进行片段大小选择,最后进行PCR富集得到最终的cDNA文库用于测序。构建原理图如下:
原核转录组建库
原核生物( 包括病毒) 的mRNA 多是多顺反子,即可以有几个基因同时被转录成一个mRNA,共同使用一个启动调控区,而真核生物多是单顺反子,即一次只转录出一个基因;原核生物mRNA与真核不同,无5′端帽子结构和3′端聚腺苷酸尾巴;所以,原核转录组建库时不能像真核生物那样用带有Oligo(dT)的磁珠富集mRNA,只能用去核糖体试剂盒(Ribo-ZeroTM kit)去除rRNA来富集mRNA。去除rRNA后其余操作步骤基本就和真核生物转录组建库一样了。
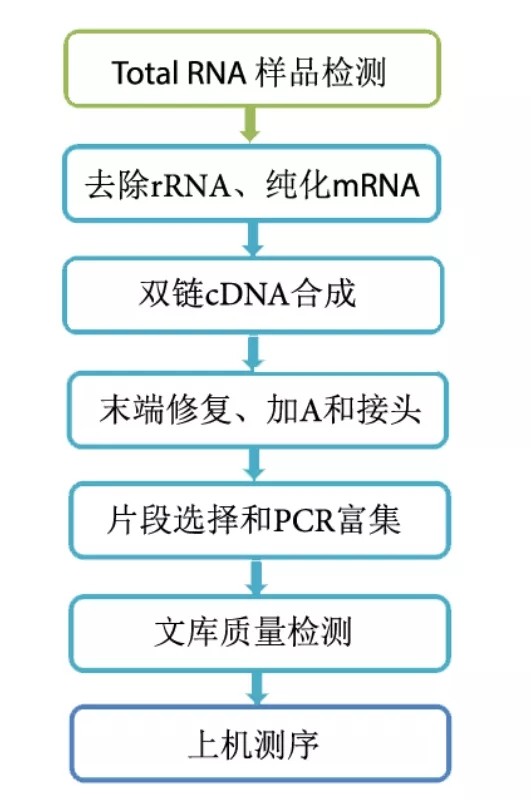
总结
以上步骤结束后就建好库了,库里面主要是片段长度在300左右的短的cDNA片段,这些片段就是来自于富集出的mRNA,这些短的cDNA片段两端都有测序接头以及barcode,以此作为身份标签,测序后可以方便地划归哪个样品。
测序出的序列称作reads,那么reads是如何转化为基因表达量信息呢?reads组装成基因吗?reads又是怎么比对到参考基因上的呢?想起来可能很简单,实际上分析过程也很复杂,想了解分析过程请持续关注本网站更新!
1. 文章越来越难发?是你没发现新思路,基因家族分析发2-4分文章简单快速,又能发文章,又能学生信技能,一举两得的SCI好思路;基因家族分析课程已更新分析内容,最新版课程学习链接:基因家族分析实操课程
2. 转录组数据结果理解不深入?图表看不懂?这就是你转录组数据不会挖掘、文章不会撰写的原因,让我带你一起深入了解转录组数据结果,学习链接:转录组(有参)结果解读;转录组(无参)结果解读
3. 转录组数据深入挖掘技能-WGCNA,提升你的文章档次,学习链接:WGCNA-加权基因共表达网络分析
4. 转录组数据怎么挖掘?多学点数据处理技能:学习链接:转录组标准分析后的数据挖掘
5.微生物多样性分析很简单,但是分析内容项目并不少,理解起来有困难?看我深入浅出讲给你听,学习链接: 微生物16S/ITS/18S分析原理及结果解读
6. 学生物的必学生信技能:Docker安装与使用、linux系统入门、Perl语言入门到精通、Python生信入门到精通、perl语言高级编程
7. 生信绘图、科研绘图技能:Cytoscape与网络图绘制、微生物OTU网络图绘制(Cytoscape)、R语言基础与绘图、R语言绘图基础(ggplot2)
8.生物信息实战技能,0基础也可以学会,内含脚本及demo数据:RNAseq有参转录组自主分析、基因组重测序自主分析、微生物多样性自主分析
9. 更多学习内容:linux、perl、R语言画图,更多免费课程请扫描下方二维码进入组学大讲堂网校学习:

- 发表于 2021-04-26 19:02
- 阅读 ( 12540 )
- 分类:转录组
