CIBERSORT R代码分析
CIBERSORT R代码分析
在R语言中运行Cibersort共需要三个文件,分别是(1)官方提供的22种细胞基因集“LM22.txt”;(2)自己的表达矩阵;(3)Cibersort代码。
(1)LM22.txt获取方法:
在Cibersort论文中(https://www.nature.com/articles/nmeth.3337#MOESM207)下载Supplementry table 1
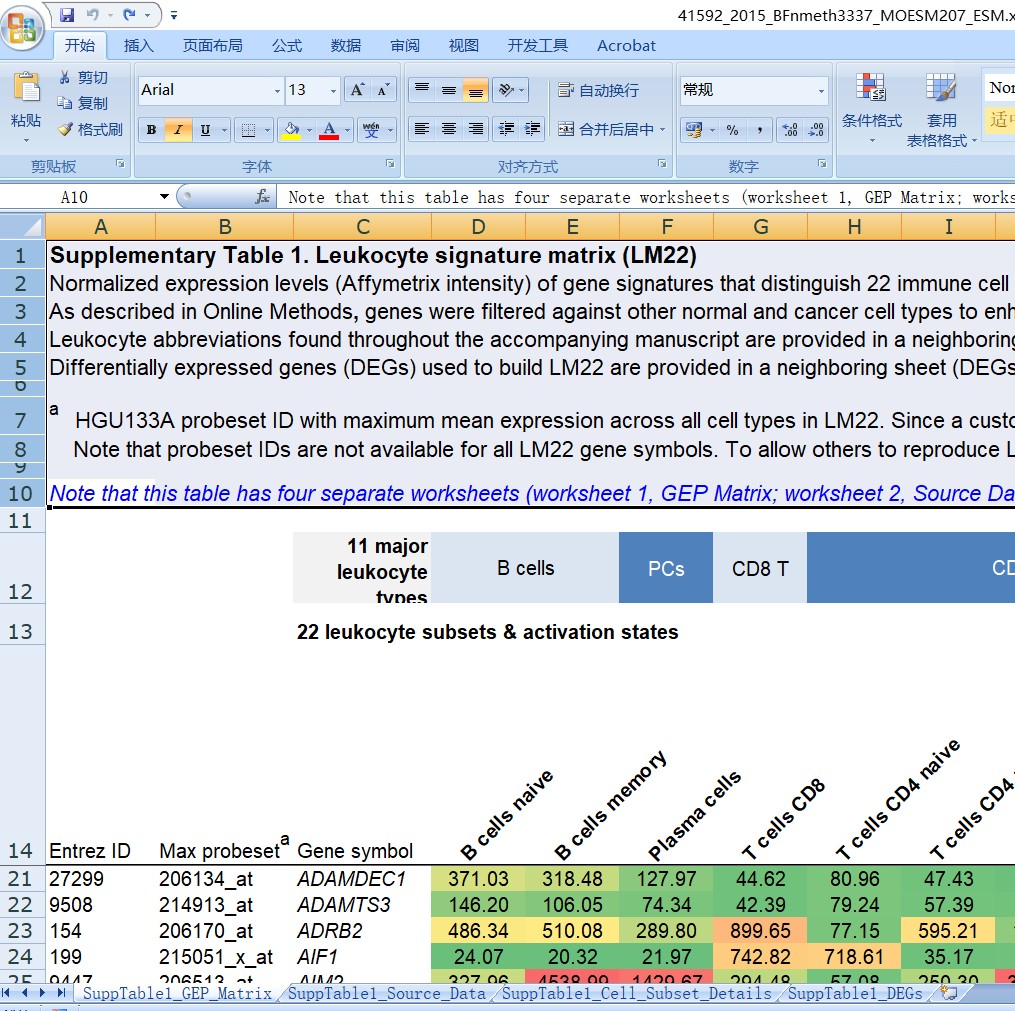
LM22.txt获取方法
删除Sheet 1 中表头,只保留矩阵部分。得到如下矩阵,另存为制表符分割的txt(“LM22.txt”)
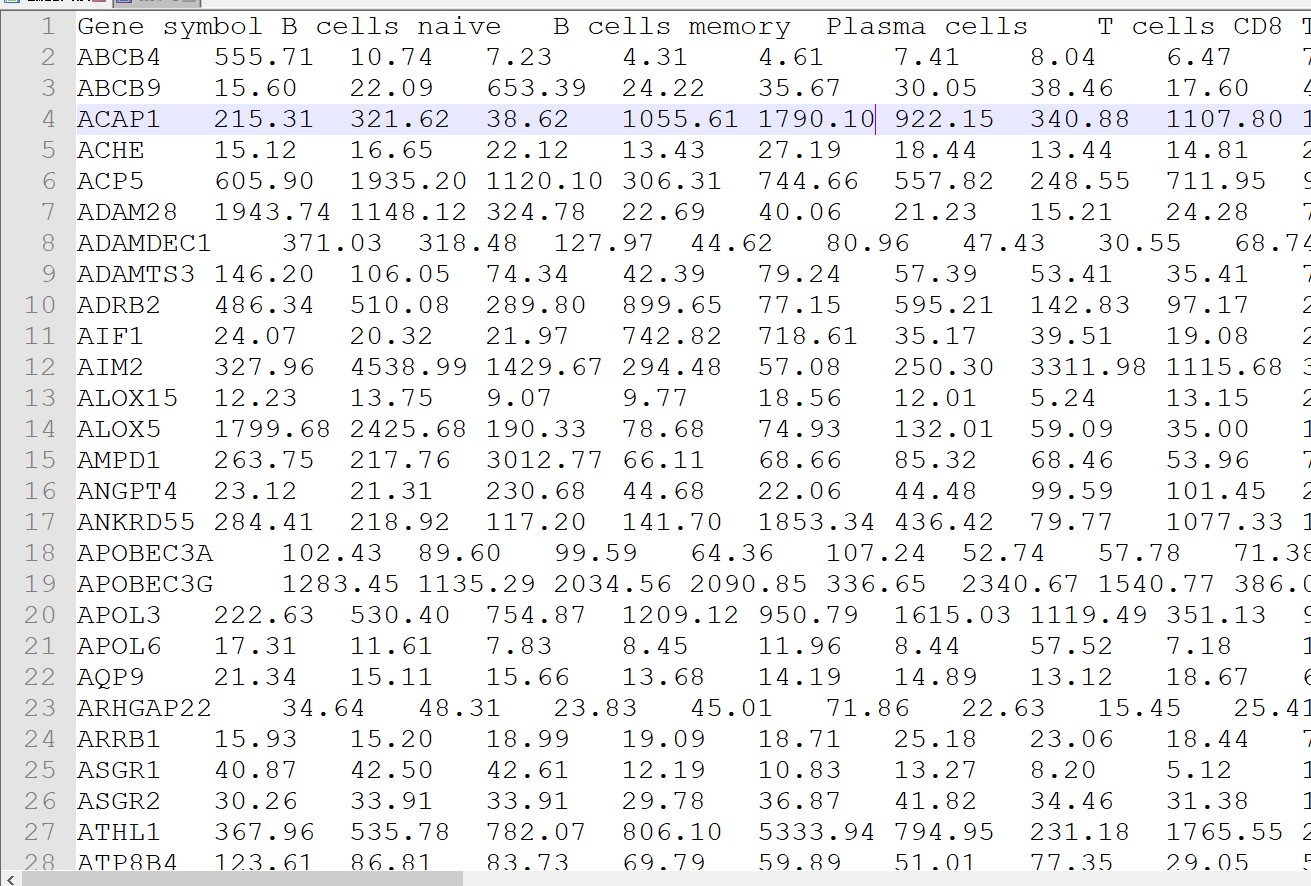
(2)自己的表达矩阵
第一列是基因名,第一行是样品名,不能有重复基因名,第一列列名不能空白。矩阵中不能存在空白或NA值,不要对表达量取Log2.
如果表达矩阵中基因不能完全覆盖LM22.txt中的基因,Cibersort同样可以正常运行,但不能少于LM22.txt中所需基因的一半。
表达矩阵保存为制表符分割的txt文本(“DATA.txt”)
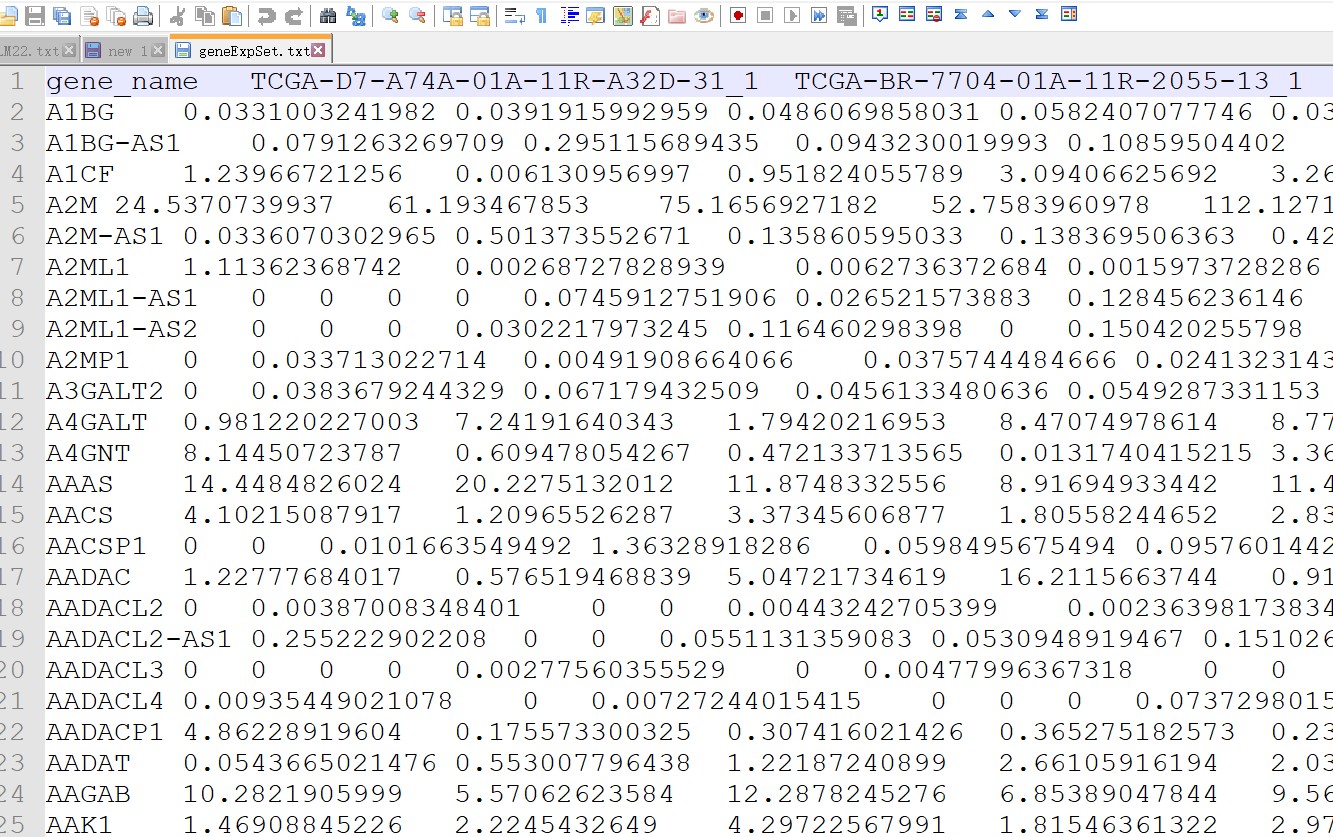
表达矩阵示例
(3)Cibersort代码
在R中新建R Script,复制以下网址中代码,保存为“Cibersort.R”
https://rdrr.io/github/singha53/amritr/src/R/supportFunc_cibersort.R
(4)以上三个文件需保存在同一文件夹,运行Cibersort的代码如下:
setwd("三个文件的文件夹")
source('Cibersort.R')
result1 <- CIBERSORT('LM22.txt','DATA.txt', perm = 1000, QN = T) #perm置换次数=1000,QN分位数归一化=TRUE
在同一文件夹下可以得到运算结果("CIBERSORT-Results.txt")
注意Cibersort结果的默认文件名为CIBERSORT-Results.txt,在同一文件夹下进行第二次运算会覆盖第一次得到的文件,建议在每一次运算之后对文件重命名。
- 发表于 2021-05-13 17:18
- 阅读 ( 6705 )
- 分类:临床医学
