enrichGO_pip.r GO富集分析
enrichGO_pip.R GO富集分析
使用说明
Rscript $scriptdir/enrichGO_pip.r -h
usage: /work/my_stad_immu/scripts/enrichGO_pip.r [-h] -g gene.list [-d ann.db]
[-t idtype] [-s show.type]
[-p pvalueCutoff]
[-q qvalueCutoff]
[-c showCategory] [-n prefix]
[-o outdir] [-H height]
[-W width]
GO enrich analysis:
optional arguments:
-h, --help show this help message and exit
-g gene.list, --gene.list gene.list
diff expressed gene list file, required
-d ann.db, --ann.db ann.db
org.Hs.eg.db or org.Mm.eg.db ,for more info visit:
https://www.omicsclass.com/article/1244 [optional,
default: org.Hs.eg.db ]
-t idtype, --idtype idtype
deg gene id type: ENSEMBL SYMBOL ENTREZID GENENAME
[optional, default: SYMBOL ]
-s show.type, --show.type show.type
set example ID type name [optional, default: ENTREZID
]
-p pvalueCutoff, --pvalueCutoff pvalueCutoff
pvalue cutoff on enrichment tests to report,
[optional, default: 0.05 ]
-q qvalueCutoff, --qvalueCutoff qvalueCutoff
qvalue cutoff on enrichment tests to report as
significant. Tests must pass i) pvalueCutoff on
unadjusted pvalues, ii) pvalueCutoff on adjusted
pvalues and iii) qvalueCutoff on qvalues to be
reported., [optional, default: 0.2 ]
-c showCategory, --showCategory showCategory
how many GO Category to show, [optional, default: 10 ]
-n prefix, --prefix prefix
the output file prefix [optional, default: enrichGO ]
-o outdir, --outdir outdir
output file directory [default cwd]
-H height, --height height
the height of pic inches [default 10]
-W width, --width width
the width of pic inches [default 10]
参数说明:
-g 输入基因列表文件: 脚本会读取第一列基因ID作为基因集:
-d 物种注释数据库: 人的:org.Hs.eg.db
-t 指定输入基因列表的ID类型
使用举例:
Rscript $scriptdir/enrichGO_pip.r --gene.list $workdir/04.deg/S1_vs_S2.DEG.final.tsv \
-o GO -n S1_vs_S2 --pvalueCutoff 0.5 --ann.db org.Hs.eg.db \
--idtype SYMBOL
结果展示:
生物学过程(biological process, BP)、分子功能(molecular function, MF)和细胞组分(cellular component, CC)
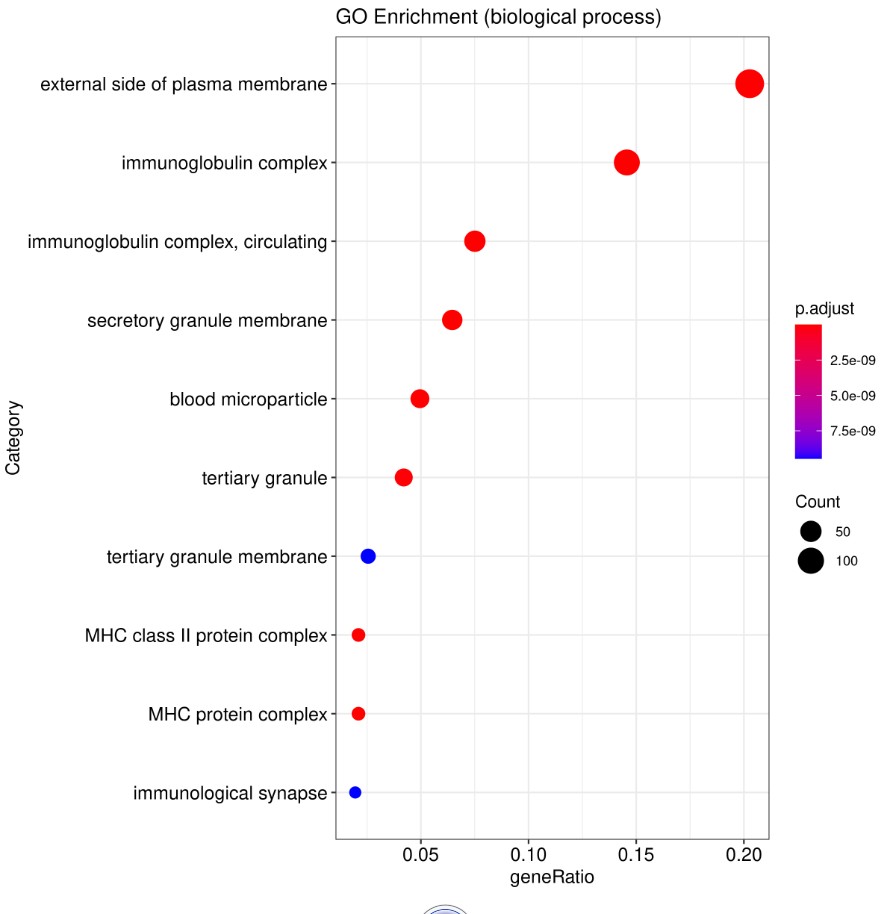
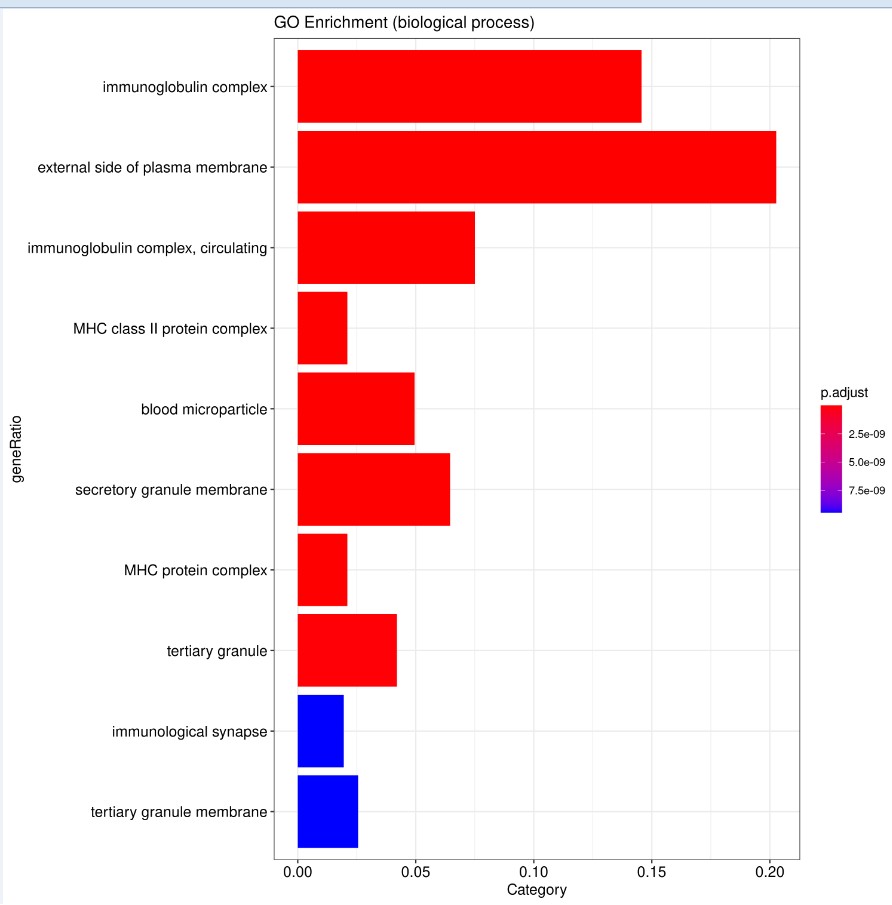
参考文献:
Yu G, Wang L, Han Y, He Q (2012). “clusterProfiler: an R package for comparing biological themes among gene clusters.” OMICS: A Journal of Integrative Biology, 16(5), 284-287. doi: 10.1089/omi.2011.0118.
- 发表于 2021-06-23 13:35
- 阅读 ( 4242 )
- 分类:转录组
