GATK外显子测序数据肿瘤体细胞突变数据分析
利用序列捕获或者靶向技术将全基因组外显子区域DNA富集后再进行高通量测序的基因组分析。外显子组包含约1%的基因组(约30MB),却包含约85%致病突变,与个体表型相关的大部分功能性变异也都集中在染色体的外显子区。能够识别单核苷酸变异体(SNVs)、小插入缺失(InDels)以及能够解释复杂遗传疾病的罕见的原发性突变。
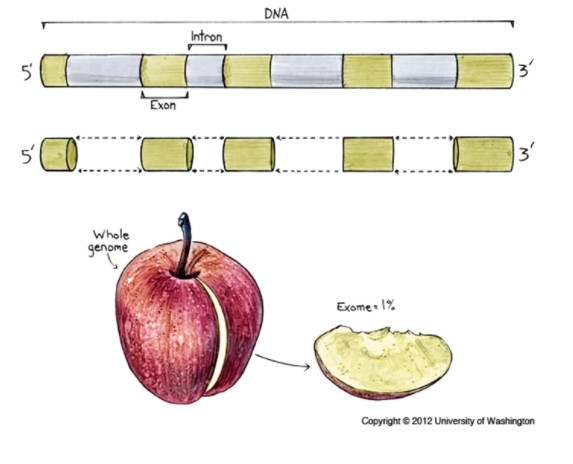
本篇文章只是分享一下利用肿瘤外显子体细胞检测分享的方法,用的工具流程参考GATK官方推荐的方法。
肿瘤数据及人类hg38参考基因组准备:
手头没有数据的可以使用GATK官网提供的示例数据,下载地址如下:
https://console.cloud.google.com/storage/browser/gatk-best-practices
hg38人类基因组相关文件GATK官方提供下载地址:
https://console.cloud.google.com/storage/browser/genomics-public-data
数据分析:
测序数据bam文件准备,按照GATK推荐的标准流程:
https://gatk.broadinstitute.org/hc/en-us/articles/360035535912
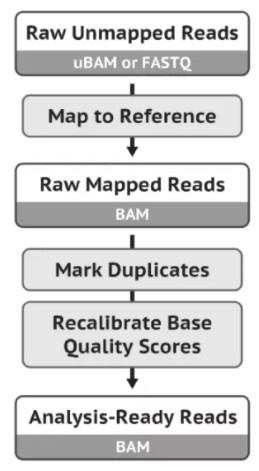
肿瘤成对样本测序的数据比对到参考基因组命令行如下:
#两个样本bwa分别比对到人类hg38参考基因组上:bwa mem Homo_sapiens_assembly38.fasta N154_1.clean.fq.gz N154_2.clean.fq.gz \ -t 8 -M -R "@RG\tID:N154\tLB:N154\tPL:ILLUMINA\tSM:N154" |samtools view -bS -h - > N154.bambwa mem Homo_sapiens_assembly38.fasta T154_1.clean.fq.gz T154_2.clean.fq.gz \ -t 8 -M -R "@RG\tID:T154\tLB:T154\tPL:ILLUMINA\tSM:T154" |samtools view -bS -h - > T154.bam
#去除PCR重复gatk MarkDuplicatesSpark -I N154.bam -O N154.sort.dedup.bam \ -M N154.sort.dedup.metrics --conf 'spark.executor.cores=4'gatk MarkDuplicatesSpark -I T154.bam -O T154.sort.dedup.bam \ -M T154.sort.dedup.metrics --conf 'spark.executor.cores=4'
#BQSR碱基质量矫正命令,分两步:gatk BaseRecalibrator -R Homo_sapiens_assembly38.fasta \ --known-sites references_hg38_v0_Homo_sapiens_assembly38.dbsnp138.vcf \ --known-sites Mills_and_1000G_gold_standard.indels.hg38.vcf.gz -I N154.sort.dedup.bam -O N154.sort.dedup.bam.tablegatk BaseRecalibrator -R Homo_sapiens_assembly38.fasta \ --known-sites references_hg38_v0_Homo_sapiens_assembly38.dbsnp138.vcf \ --known-sites Mills_and_1000G_gold_standard.indels.hg38.vcf.gz \ -I T154.sort.dedup.bam -O T154.sort.dedup.bam.table
#print readsgatk ApplyBQSR -R Homo_sapiens_assembly38.fasta \ -I N154.sort.dedup.bam --bqsr-recal-file N154.sort.dedup.bam.table \ -O N154.sort.dedup.bqsr.bamgatk ApplyBQSR -R Homo_sapiens_assembly38.fasta \ -I T154.sort.dedup.bam --bqsr-recal-file T154.sort.dedup.bam.table \ -O T154.sort.dedup.bqsr.bam
2.体细胞突变检测
bam文件准备好之后用GATK中Mutect2工具做体细胞突变检测:
https://gatk.broadinstitute.org/hc/en-us/articles/360035894731-Somatic-short-variant-discovery-SNVs-Indels-
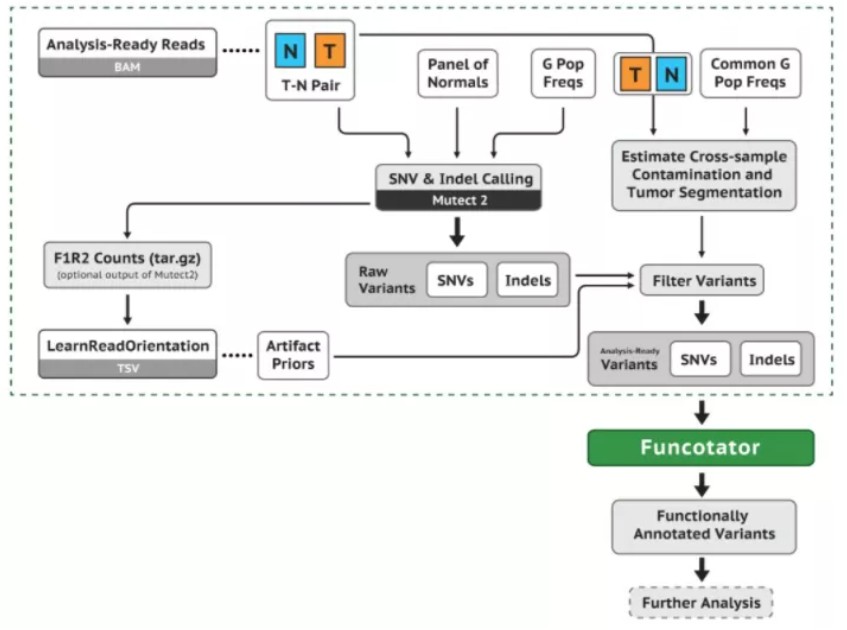
#Mutect2 体细胞检测gatk Mutect2 -R Homo_sapiens_assembly38.fasta \ -I N154.sort.dedup.bqsr.bam -I T154.sort.dedup.bqsr.bam \ -normal N154 --panel-of-normals somatic-hg38_1000g_pon.hg38.vcf.gz \ --germline-resource somatic-hg38_af-only-gnomad.hg38.vcf.gz \ -L S33613271_Regions.bed -O 154.raw.somatic.vcf.gz#-L 参数可以指定捕获区域#--panel-of-normals 指定正常人的突变信息
#质控过滤gatk FilterMutectCalls -R Homo_sapiens_assembly38.fasta \ -V 154.raw.somatic.vcf.gz -O 154.filter.checked.vcf.gz &&\ zcat 154.filter.checked.vcf.gz |awk '$0~/#/ || ($7 =="PASS"){print $0}' |bgzip >154.clean.vcf.gz && rm -f 154.filter.checked.vcf.gz
3.对突变结果进行注释
使用的软件为ANNOVAR,注释命令如下,需要整理人类注释文件humandb/hg38/可到ANNOVAR官方网站下载:
https://annovar.openbioinformatics.org/en/latest/user-guide/download/
命令行如下:
table_annovar.pl 154.clean.vcf.gz humandb/hg38/ \ -buildver hg38 -out 154 -remove -protocol refGene,cosmic70,nci60,esp6500siv2_all,clinvar_20210501,1000g2015aug_all,1000g2015aug_eas,1000g2015aug_sas,avsnp150,gwasCatalog,ljb26_all,cytoBand,dgvMerged,phastConsElements100way,genomicSuperDups -operation g,f,f,f,f,f,f,f,f,f,f,r,r,r,r -nastring . -vcfinput
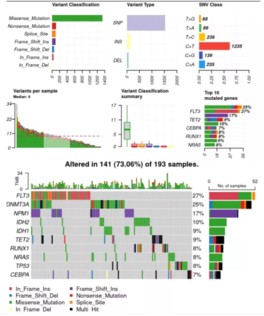
4.体细胞突变结果可视化
利用maftools工具对结果进行可视化展示:
#将ANNOVAR注释后的结果整理成maftools要求的结果格式 for i in *.hg38_multianno.txt;do sample=`echo $i|awk -F '.' '{print $2}'` cut -f '1-10' $i|sed '1d'|sed "s/$/\t${sample}/">>all_sample.txtdonesed -i '1s/^/Chr\tStart\tEnd\tRef\tAlt\tFunc.refGene\tGene.refGene\tGeneDetail.refGene\tExonicFunc.refGene\tAAChange.refGene\tTumor_Sample_Barcode\n/' all_sample.txt
# R代码汇总绘图,读入数据并可视化绘图library(maftools)var_maf= annovarToMaf(annovar = "all_sample.txt", Center = 'NA', refBuild = 'hg38', tsbCol = 'Tumor_Sample_Barcode', table = 'refGene',MAFobj =T, sep = "\t")
plotmafSummary(maf = var_maf, rmOutlier = TRUE, addStat = 'median')oncoplot(maf = var_maf, top = 30, fontSize = 12 ,showTumorSampleBarcodes = F )
参考文献:
Ng SB1, Turner EH., et al. Targeted capture and massively parallel sequencing of 12 human exomes. Nature.461(7261):272-6.
ChoiM1,Scholl UI., et al. Genetic diagnosis by whole exome capture and massively parallel DNA sequencing.Proc Natl Acad Sci USA.106(45):19096-101.
- 发表于 2022-02-11 16:14
- 阅读 ( 5625 )
- 分类:重测序
