IF=5.6|GBM单细胞数据挖掘实践
TCGA数据挖掘已经烂大街, 单细胞与空间转录组数据分析近年来十分火热,公开的数据也越来越多, 如果结合单细胞数据做生信挖掘分析发文相对新颖且容易得多,接下来我们介绍相应的挖掘文章思路,再利用我们提供的代码可以轻松复现文章中单细胞分析部分.
1.GBM单细胞数据挖掘文献介绍

1.1文章数据挖掘思路:
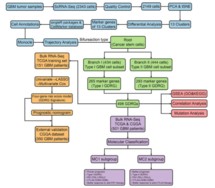
1.2 单细胞分析部分主要结果:
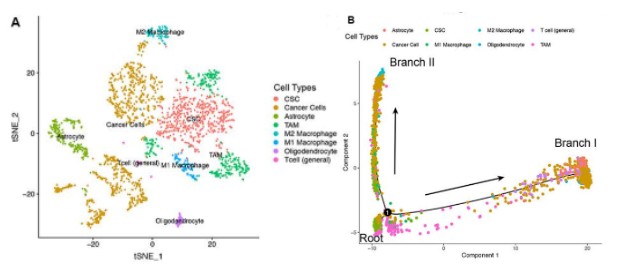
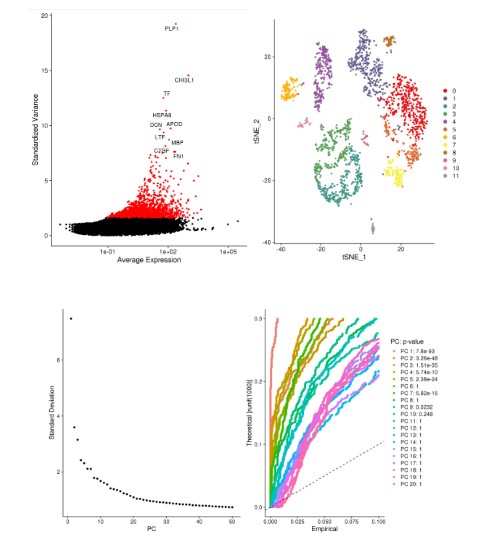
单细胞数据集来自GEO数据:GSE84465, 其中4个GBM样本共3589个单细胞表达数据,作者筛选其中的2343个来源于肿瘤核心部位的细胞进行分析。分析用到的R包为Seurat 。经过质控及标准化数据,194个低质量细胞被过滤掉).总共纳入19752个基因分析,方差分析找到1500个高变的基因。之后,tSNE分析将GBM细胞分为13个亚群,差异表达分析从所有13个亚群中鉴定出总共8025个标记基因。根据标记基因的表达量,我们使用singleR和CellMarker对各个亚群进行细胞类型注释.
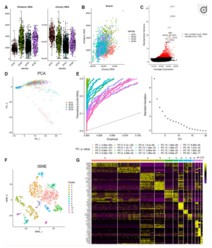
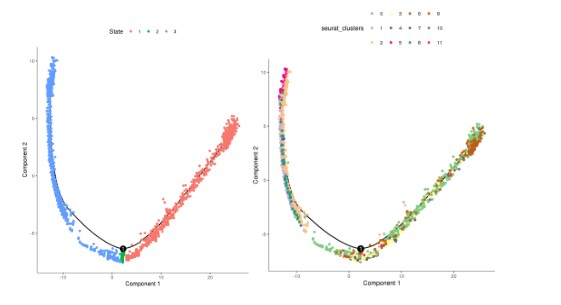
使用Monocle 2算法进行时序及轨迹分析并确定GBM分化相关基因( GBM cell differentiation-related genes GDRG ). 拟时序分析结果发现肿瘤干细胞主要位于树根部(root),而2个树枝(branch)分布有不同的GBM细胞,branch I包含了434个GBM细胞(下图A),而branch II包含了444个细胞(下图B)。差异表达分析得到265个I型GDRG和193个II型GDRG。基因集富集分析(GSEA)发现I型GDRG与免疫反应调控相关通路显著负相关,而II型GDRG与代谢相关通路显著正相关.
筛选到GDRG基因之后,再结合TCGA和CGGA当中的数据进一步筛选相关的预后基因,再构建预后模型,为常规套路, 我们这里主要介绍一下如何筛选到GFRG;
2.单细胞数据分析复现代码讲解
数据下载: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE84465
2.1 单细胞表达矩阵下载
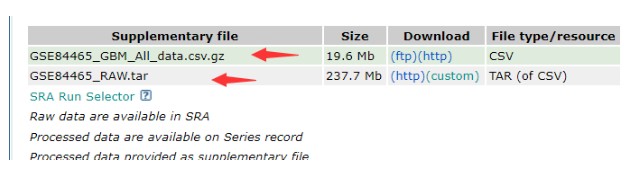
两个文件都需要下载,两个文件都是细胞表达矩阵,我们主要用第一个文件,打开之后显示,列为细胞ID,行为基因.中间为不同的基因在不同的细胞中 的表达量;
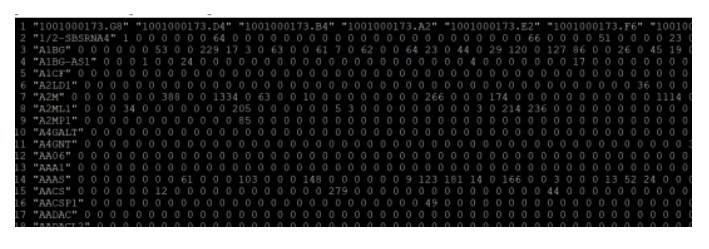
2.2 细胞信息metadata文件准备:
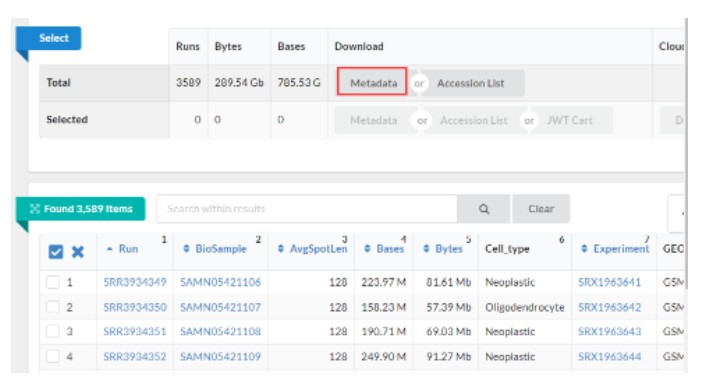
首先这个地址下载每个细胞的信息表格https://www.ncbi.nlm.nih.gov/Traces/study/?acc=SRP079058
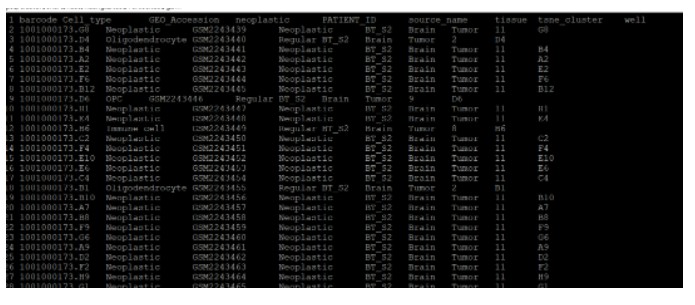
其次, 由于我们的表达矩阵细胞ID是不在这个meta表中,我们需要利用GSE84465_RAW.tar这个文件找到 GEO_Accession 与细胞ID之间的对应表格,整理完成之后如下 文件 gbm.meta.tsv :
2.3 数据读入与过滤:
作者是使用tumor cores 细胞做分析,因此需要对metadata文件gbm.meta.tsv进行筛选, 筛选倒数第三列为Tumor的的行保留,即保留了肿瘤细胞做分析:

然后按照作者方法部分读入数据并过滤数据, 代码如下:
#筛选肿瘤细胞用于分析 awk -F"\t" 'NR==1|| $7=="Tumor"{print}' gbm.meta.tsv >gbm.meta.Tumor.tsv #数据读入与质控R代码一键完成 Rscript scripts/seurat_sc_qc.r -i GSE84465_GBM_All_data.tsv.gz \ --metadata gbm.meta.Tumor.tsv --project GBM -p gbm --sep " " -o 01.read.gbm \ --nGene.min 50 --percent_mito 5
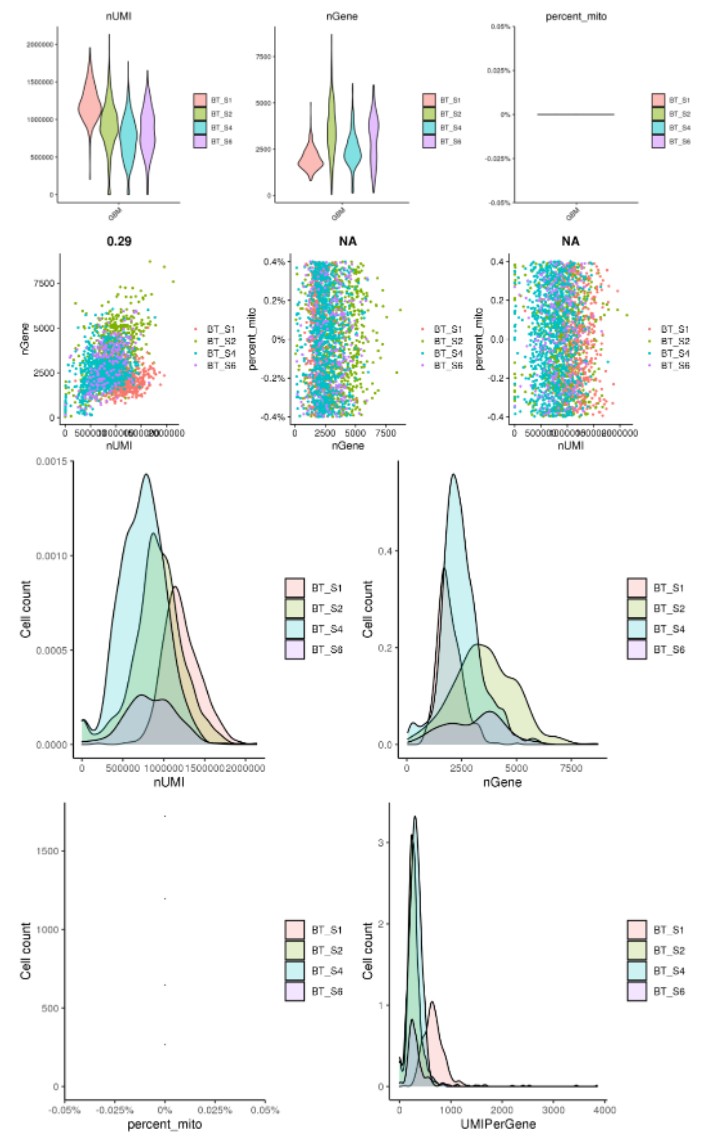
代码运行完成结果展示, 不同样本基因数量分布,UMI分布,线粒体分布等:
2.4 聚类分析:
作者方法中说明的参数较少,不能完全复现文章内容,但是整体结果还是一致的

代码一键完成:
Rscript scripts/seurat_sc.r --resolution 0.4 --dim 20 --rds 01.read.gbm/gbm.afterQC.rds -o 02.cluster -p gbm结果展示:
2.5 查找marker基因
Rscript scripts/seurat_FindAllMarkers.r -i 02.cluster/gbm.rds -o 02.cluster -p gbm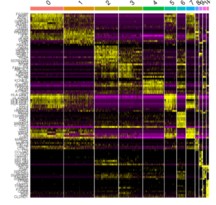
2.6 拟时序分析,
可以得到每个细胞的发育轨迹,与状态分组:
2.7 查找GDRG
其实就是将细胞状态分组(1,2,3) 添加到metadata中,之后就可以用findMarkers筛选特定细胞状态的特征基因 GDRG.
#查找特异marker基因# mkdir 04.findMarker #提取状态信息 awk -F'\t' 'BEGIN{OFS="\t" }{print $1,$26,$27}' 03.trajectory/gbm.Pseudotime.tsv >04.findMarker/Pseudotime.tsv #添加拟时序信息分组添加到seurat对象的metadata中 Rscript scripts/addMetadata_to_seurat_obj.r -i 02.cluster/gbm.rds \ -m 04.findMarker/Pseudotime.tsv -o 04.findMarker -p gbm.Pseudotime
#差异基因/MARKER基因查找 stat1 Rscript scripts/seurat_FindMarkers.r -i 04.findMarker/gbm.Pseudotime.rds \ --ident.1 1 --group.by State -o 04.findMarker -p State1.marker
#差异基因/MARKER基因查找 stat3 Rscript scripts/seurat_FindMarkers.r -i 04.findMarker/gbm.Pseudotime.rds \ --ident.1 3 --group.by State -o 04.findMarker -p State3.marker
总结:
利用代码命令行进行数据分析, 可批量重复分析数据, 大大提高数据分析效率;由于文章中方法部分写的信息不是很全面,某些参数使用可能不一样,导致我们分析的结果可能与文章中有细微差异,但是 不影响结果总体的一致性
参考文献
Wang Z, Guo X, Gao L, Wang Y, Ma W, Xing B. Glioblastoma cell differentiation trajectory predicts the immunotherapy response and overall survival of patients. Aging (Albany NY). 2020 Sep 21;12(18):18297-18321. doi: 10.18632/aging.103695. Epub ahead of print. PMID: 32957084; PMCID: PMC7585071.
延伸阅读- GEO数据库挖掘—WGCNA鉴定骨肉瘤转移相关基因
- GEO、TCGA多数据库联合挖掘胰腺导管腺癌预后关键基因
- 文献精读-GEO数据挖掘生物信息文章(宫颈癌)
- GEO数据挖掘直肠
- GEO数据挖掘案例解读-植物篇(拟南芥重金属)
- GEO数据如何挖掘?案例解析!
- 胃癌免疫侵润预后Signature数据挖掘-糖酵解
- IF:8.8|TCGA+ceRNA+肝癌文献解析
- IF=8.78|非肿瘤数据挖掘思路(GWAS Catalog)
- 发表于 2022-05-26 09:33
- 阅读 ( 6445 )
- 分类:转录组
