Infercnv|单细胞转录组拷贝数分析
inferCNV用于探索肿瘤单细胞RNA-seq数据,分析其中的体细胞大规模染色体拷贝数变化(copy number alterations, CNA),例如整条染色体或大片段染色体的增加或丢失(gain or deletions)。工作原理是,以一组“正常”细胞作为参考,分析肿瘤细胞基因组上各个位置的基因表达量强度变化,通过热图的形式展示每条染色体上的基因相对表达量,相对于正常细胞,肿瘤细胞基因组总会过表达或者低表达。
文章案例
2021年5月发表在Nat Commun上的一篇文章《Single-cell profiling of tumor heterogeneity and the microenvironment in advanced non-small cell lung cancer》(IF=14.92)中使用scRNA-seq数据来推断癌细胞群中的拷贝数改变 (CNA)。通过CNV图谱推断患者间和患者内的异质性。
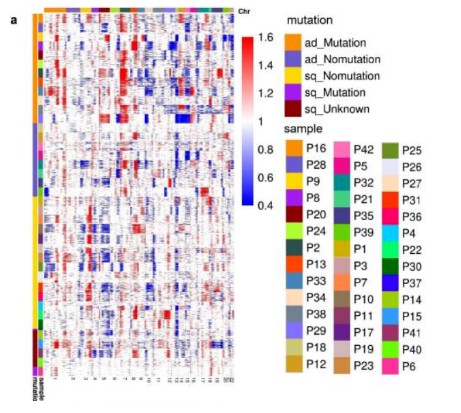
拷贝数分析
为了使大家能更简便快捷地对单细胞数据进行拷贝数分析,我们给大家提供了一个R脚本。使用该脚本只需要准备好相应的输入文件,再进行简单的命令行操作即可进行拷贝数分析并绘制可直接用于文章发表的图片。
使用命令:
Rscript single_cell/infercnv.r -i data/cnv/merged.rds --downsample 100 -g 0 1 2 -o cnv_result
结果展示:

输入文件准备:本脚本所必需的输入文件只有一个,即经聚类注释后的seurat对象,为rds文件。
更多脚本参数设置及说明,通过-h参数获得以下帮助信息:
usage: single_cell/infercnv.r [-h] -i rds_data [--assay assay]
[--downsample downsample] [--species species]
[-t type] -g group [group ...]
[--platform platform] [--hmm] [-o path]
single cell trajectory analysis : http://cole-trapnell-lab.github.io/monocle-
release/docs/#constructing-single-cell-trajectories
optional arguments:
-h, --help show this help message and exit
-i rds_data, --rds_data rds_data
input rds_data file path[required]
--assay assay Name of assay to use, defaults to the active assay
[default RNA]
--downsample downsample
subset cells numbers for analysis [default None]
--species species Select the species. Available species are:homo(Homo
sapiens),mus(Mus musculus) [default homo]
-t type, --type type the type of cell [default seurat_clusters]
-g group [group ...], --group group [group ...]
group for ref cell type [required]
--platform platform Select type of Single cell sequencing platform.
Available types are:10x-genomics,smart-seq [default
10x-genomics]
--hmm Not hmm predict cnv [default False]
-o path, --outdir path
output file directory [default
/share/nas5/zhangll/self/single_cell]
必需参数:
-i 经聚类注释后的seurat对象,为rds文件
-g 参考细胞类群,类群名字应属于-t参数所指定组别内
其他参数:
-t 细胞注释类别列名
--assay 指定seurat对象中的assay,默认为RNA--downsample 是否减少样本量,默认为False
--species 所分析物种,可选物种包括homo(Homo sapiens)、mus(Mus musculus),默认为homo
--platform 数据来源所属平台,可选平台包括10x-genomics、smart-seq,默认为10x-genomics
--hmm 是否根据hmm模型推测cnv,默认为False
-o 输出路径,默认为当前路径
脚本获取方法
为了方便感兴趣的学员小伙伴想复现这个分析内容,我把数据和代码一并打包,免费供大家使用学习。只需要如下几步,即可获得下载链接。
1、关注生信博士公众号;
2、转发本文章至朋友圈,并截图,在公众号菜单栏对话框发送截图;
3、在公众号菜单栏回复infercnv即可获得单细胞cnv分析及相关图的绘制脚本的网盘链接以及提取码。
如果没有数据分析的基础,不知道从何下手,可以学习一下我们的《医学数据分析环境搭建》视频课程,扫描下方二维码了解课程详情。

延伸阅读
- GEO数据库挖掘—WGCNA鉴定骨肉瘤转移相关基因
- GEO、TCGA多数据库联合挖掘胰腺导管腺癌预后关键基因
- 文献精读-GEO数据挖掘生物信息文章(宫颈癌)
- GEO数据挖掘直肠
- GEO数据挖掘案例解读-植物篇(拟南芥重金属)
- GEO数据如何挖掘?案例解析!
- 胃癌免疫侵润预后Signature数据挖掘-糖酵解
- IF:8.8|TCGA+ceRNA+肝癌文献解析
- IF=8.78|非肿瘤数据挖掘思路(GWAS Catalog)

- 发表于 2022-06-10 10:14
- 阅读 ( 4117 )
- 分类:转录组
