Logistic回归模型介绍及使用(附脚本)
回归模型是统计中最常用的模型之一,它主要用于解释和预测。本文要讲解一种应用非常广泛的回归模型:Logistic回归(逻辑回归)。
Logistic回归方法主要是针对二分类问题的分析,即被预测的变量为二元变量,如成功或失败、流失或不流失等,常用于机器学习与生信分析中。
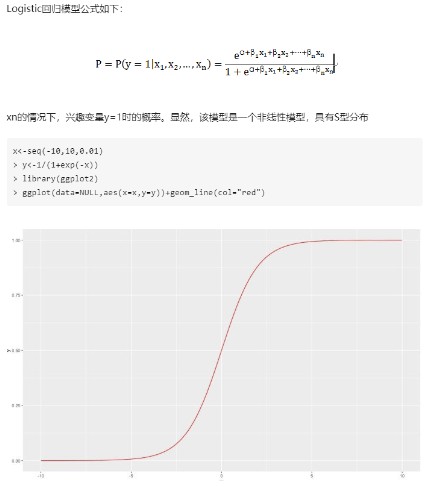
为了方便大家能更加高效快捷地使用Logistic回归模型对自身数据进行建模,这里我们给大家提供了一个构建Logistic回归模型的R脚本。使用该脚本都只需要准备好相应的输入文件,再进行简单的命令行操作即可。
使用命令示例如下
Rscript /share/work/fangs/scripts/tcga/logic_batch.r -m metadata_all.tsv -g immu_nes.tsv -v Angiogenesis APM1 B_cell_PCA_16704732 Bcell_receptors_score Buck14_score CD103neg_mean_25446897 CD103pos_mean_25446897 CD8_PCA_16704732 CHANG_CORE_SERUM_RESPONSE_UP CSF1_response -B barcode -e EVENT -l 0.05 -p immu
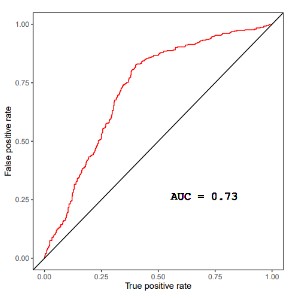
上述命令分别会得到两个结果文件以及ROC曲线图。
一个是logistic回归的结果文件:第一列为变量(Variable)、第二列为每个变量相关的β系数估计值(Estimate)、第三列为系数估计值的标准误(Std.Error)、第四列为Z统计量,即系数估计值除以估计值的标准误(Z)、第五列为对应于Z统计量的显著性P值(P)。如下表所示:
| Variable | Estimate | Std.Error | Z | P |
| CD103neg_mean_25446897 | 7.72727446885497 | 2.13196826481262 | 3.62447912400522 | 0.000289544405375361 |
| CD103pos_mean_25446897 | -4.99678794955958 | 1.38255892114382 | -3.6141591314066 | 0.00030132395784459 |
| CHANG_CORE_SERUM_RESPONSE_UP | -9.15989987434626 | 3.00322462051504 | -3.05002157073931 | 0.00228824930868181 |
| Buck14_score | 4.70034740314082 | 1.83094184032087 | 2.56717460906191 | 0.0102530968385302 |
另一个也是logistic回归的结果文件,与上个结果文件不同的是通过显著性P值对变量进行了筛选,只保留了显著与被预测变量(结果变量)相关的变量。
ROC曲线图:
输入文件准备:
这个脚本必需的输入文件有两个:
一个为样本信息文件(metadata_all.tsv),通过-m参数指定。这个文件中必须含有样本编号(barcode)及被预测变量(EVENT)两列,我们这里的被预测变量是患者的生存状态(0为存活,1为死亡)。下表为部分文件内容展示:
| barcode | TIME | EVENT |
| TCGA-E2-A158-11A-22R-A12D-07 | 450 | 0 |
| TCGA-BH-A0DD-11A-23R-A12P-07 | 2486 | 0 |
另一个输入文件通过-g参数指定(immu_nes.tsv)。这个文件是变量在每个样本中的值(行为变量,列为样本),若变量是基因,那么就是基因表达量文件。这里我们输入的是ssgsea分析富集所得的结果文件(行为特征基因类型,列为样本)。下表为部分文件内容展示:
| SetName | TCGA-E2-A158-11A-22R-A12D-07 | TCGA-BH-A0DD-11A-23R-A12P-07 |
| Angiogenesis | 0.726968114703625 | 0.732888990014272 |
| APM1 | 0.724390452299456 | 0.731456809304532 |
更多脚本参数设置及说明
初次使用脚本时,可以通过-h参数获得以下帮助信息。
Rscript /share/work/fangs/scripts/tcga/logic_batch.r -h
usage: /share/work/fangs/scripts/tcga/logit_batch_test.r [-h] -m metadata -g
expset [-B by]
[-e event] -v variate
[variate ...]
[-l pvalue] [--log2]
[-o outdir]
[-p prefix]
[-H height]
[-W width]
batch unvariate cox regression gene expression:
https://www.omicsclass.com/article/1507
optional arguments:
-h, --help show this help message and exit
-m metadata, --metadata metadata
input metadata file path with suvival time[required]
-g expset, --expset expset
input gene expression set file[required]
-B by, --by by input sample ID column name in metadata[default
barcode]
-e event, --event event
set event column name in metadata[default EVENT]
-v variate [variate ...], --variate variate [variate ...]
variate for cox analysis[required]
-l pvalue, --pvalue pvalue
pvalue cutoff to choose sig gene[default 0.05]
--log2 whether do log2 transfrom for expression
data[optional,default: False]
-o outdir, --outdir outdir
output file directory[default cwd]
-p prefix, --prefix prefix
out file name prefix[default cox]
-H height, --height height
the height of pic inches[default 5]
-W width, --width width
the width of pic inches[default 5]
-B 指定两个输入文件合并的关键列的列名,这里根据样本名合并故指定为barcode,需要注意的是两个文件中的样本名必须相同;
-e 指定被预测变量的列名,需为二分类变量;
-v 指定变量的名字,需与第二个输入文件中变量的名字相同;
-l 指定显著性P值的阈值,这里认为P<0.05则显著;
--log2 加入该参数则对第二个文件中变量的值进行log2转换。
其他参数说明
-H 指定输出图片的高度,默认高为6英寸;
-W 指定输出图片的宽度,默认宽为8英寸;
-o 指定结果输出的路径,默认为当前路径;
-p 指定输出文件名字的前缀。
脚本获取方法
为了方便感兴趣的学员小伙伴想复现这个分析内容,我把数据和代码一并打包,免费供大家使用学习。只需要如下几步,即可获得下载链接。
1、关注生信博士公众号;
2、转发本文章至朋友圈,并截图,在公众号菜单栏对话框发送截图;
3、在公众号菜单栏回复logic即可获得单细胞cnv分析及相关图的绘制脚本的网盘链接以及提取码。
如果没有数据分析的基础,不知道从何下手,可以学习一下我们的《医学数据分析环境搭建》视频课程,扫描下方二维码了解课程详情。

- 发表于 2022-06-10 15:29
- 阅读 ( 4097 )
- 分类:R
