Clustal Omega多序列比对使用方法(命令行及在线网站)
多序列比对在保守区域鉴定,系统发育分析,motif识别等多个领域发挥重要作用,是生物信息数据分析必备的基础技能之一。Clustal是一款经典的多序列比对工具,支持DNA, RNA, 蛋白质的比对。官网如下http://www.clustal.org/
Clustal 有两个版本可用,之前的版本同时提供了GUI和命令行两种工具,GUI版的叫做ClustalX, 命令行版叫做ClustalW; 最新版本叫做Clustal Omega, 提供了命令行版,还可以通过在线服务网址进行可视化操作。
Clustal Omega是欧洲生物信息研究所(EBI)开发的多序列比对排列工具,现已经完全取代了之前ClustalW的地位。最新本的Clustal omega比对准确度更高,而且速度更快,适合大规模的多序列比对。使用该工具不仅能够对DNA或者蛋白质进行多序列比对,并且可以自动生成多种格式或构建进化树等。经测试,Clustal omega大规模多序列比对的速度比Muscle更快。

一、命令行操作
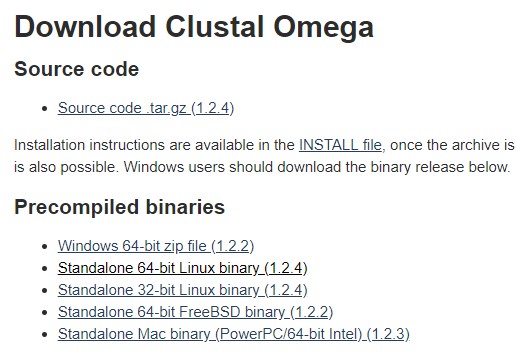
1.软件下载
在官网上,提供了源代码和编译好的二进制文件,通常情况下,直接下载对应的二进制可执行文件就行了。
网址为:https://www.ebi.ac.uk/Tools/msa/clustalo/
2.软件的基本用法
clustalo -i seq.fasta > align.fa
-i 指定输入的序列文件,默认输出结果打印在屏幕上,可以重定向到指定文件中。该软件支持多种格式的输出(fasta, clustal, msf, phylip, selex, Stockholm, Vienna)。默认输出格式为fasta, 可以通过--outfmt参数指定输出文件的格式。多序列比对不同于Blast的地方在于,Blast是局部比对,而多序列比对是全局比对。全局比对意味着需要将输入序列对齐到同一个水平来比对,一般是通过在输入序列中插入-来使得序列对齐。
二、在线服务网址操作
如果不习惯命令行的操作方式,也有在线服务可以使用。EBI提供的在线服务网址为:https://www.ebi.ac.uk/Tools/msa/clustalo/
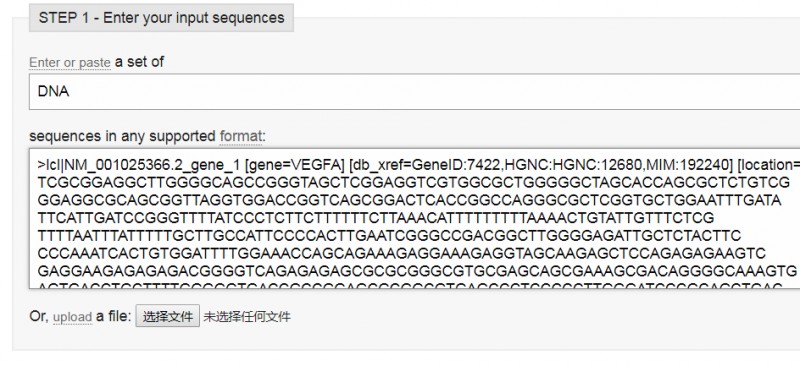
1. 打开该网页,选择正确的序列类型,将之前得到的DNA或蛋白质序列全部复制、粘贴到序列框中(或上传序列文件)。
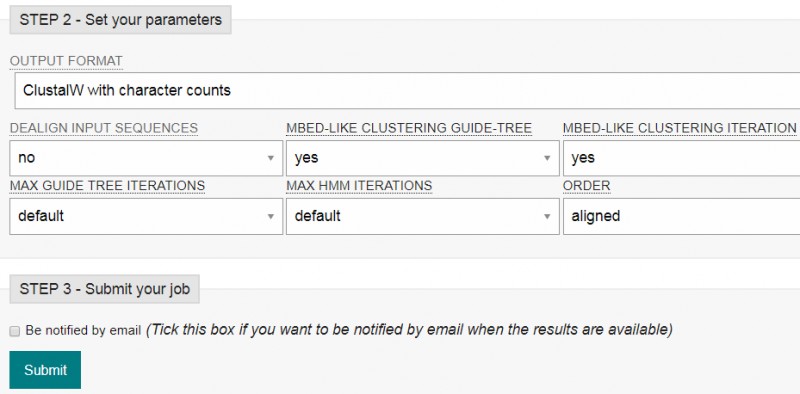
2. 参数设置推荐默认就好,点击Submit:
3. 经过一段时间等待,出现以下结果:
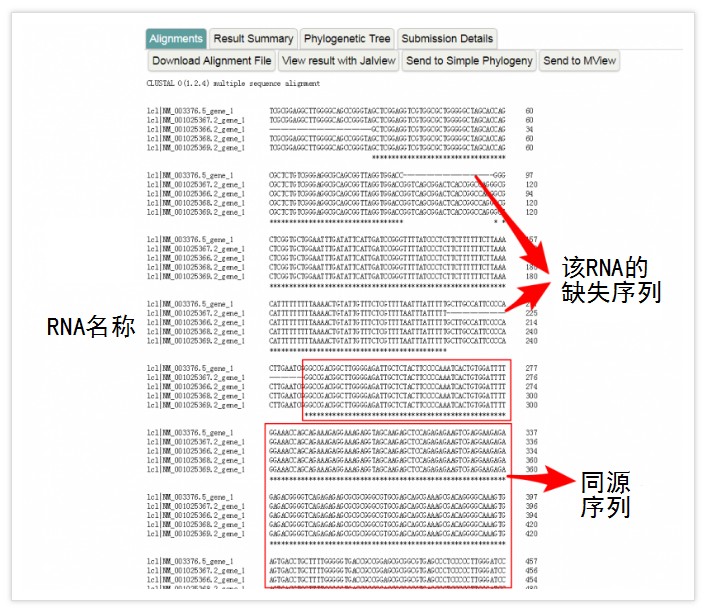
该结果将多个RNA按照同源序列重新进行排列,其中,共有序列下方以*表示,而非同源区域则以--隔开。
4. 定位共有序列的区域
这样,在比对结果中找到连续的*所对应的位置(一定要连续的),就是这几个转录变体的共有序列所在区域。
但是这种比对形式无法直接得到序列,可以将多行共有序列一一复制粘贴拼接在一起,也可以在任意一个转录变体中搜索共有序列的头和尾一小段,中间的就是共有序列。
此外,在输出结果中,还提供了颜色标记,进化树可视化等功能。
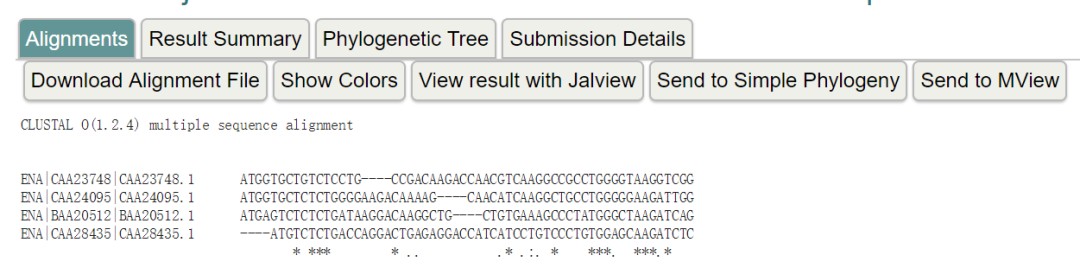
通过Mview可视化多序列比对结果;也支持导出到Jalview软件中进行可视化。
通过Phylogenetic Tree可以查看进化树的结果,默认采用NJ法建树,示意如下也可以通过Send to Simple Phylogeny, 创建进化树,支持NJ和UPGMA两种建树方式。
参考: https://blog.csdn.net/weixin_43569478/article/details/108079224
- 发表于 2022-09-09 14:21
- 阅读 ( 38074 )
- 分类:软件工具
