Perl方法对文档全文进行字符串对应替换
举个例子:从数据库中下载基因组数据,基因组.gff文件中染色体ID较为复杂(第一列)
从全基因组序列.genome.fa文件中找到染色体ID对应简写
想将gff文档中染色体ID全部替换成第二列LC*命名...
举个例子:从数据库中下载基因组数据,基因组.gff文件中染色体ID较为复杂(第一列)
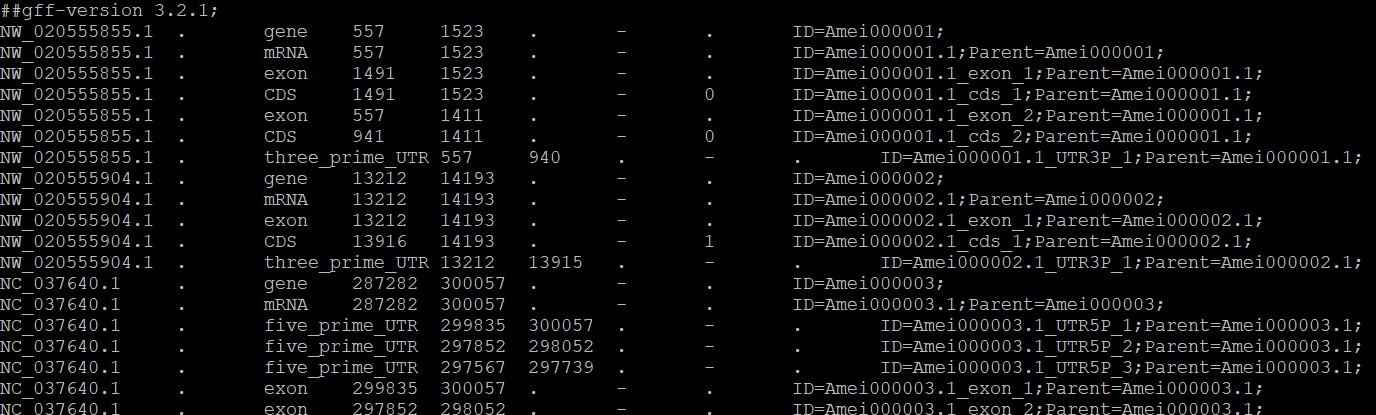
从全基因组序列.genome.fa文件中找到染色体ID对应简写
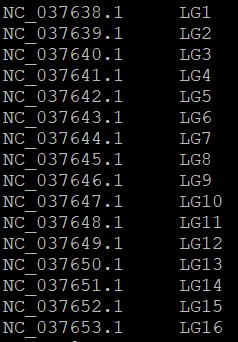 想将gff文档中染色体ID全部替换成第二列LC*命名
想将gff文档中染色体ID全部替换成第二列LC*命名
use Data::Dumper;
use Getopt::Long;
use strict;
use Cwd qw(abs_path getcwd);
my %opts;
GetOptions (\%opts,"idlist=s","gff=s","out=s");
if(!defined($opts{idlist})||!defined($opts{gff})||!defined($opts{out})){
&USAGE;
}
######################get gff##############################
my $gff;
open(IN,"$opts{gff}")||die "open gff failed\n";
{
local $/=undef;
$gff=<IN>;
close(IN);
}
#####################get idlist for changing###############
open(IN,"$opts{idlist}")|| die "open ID file failed\n";
while(<IN>){
chomp;
my($newID,$gene)=split(/\s+/,$_);
if($gff=~/$gene/){
$gff=~ s/$gene/$newID/g;
}
}
close(IN);
#####################print out new gff#######################
open(OUT, ">$opts{out}")||die "open new gff failed\n";
print OUT $gff;
close(OUT);
sub USAGE{
print "usage:perl gffID.pl -idlist idlist.txt -gff genome.gff -out rename.gff \n";
exit;
}
- 发表于 2022-10-24 14:38
- 阅读 ( 1663 )
- 分类:perl
